library(tidyverse)
library(palettetown)
library(ggridges)
library(patchwork)
library(brms)
library(bayesplot)
library(tidybayes)19 Metric Predicted Variable with One Nominal Predictor
This chapter considers data structures that consist of a metric predicted variable and a nominal predictor…. This type of data structure can arise from experiments or from observational studies. In experiments, the researcher assigns the categories (at random) to the experimental subjects. In observational studies, both the nominal predictor value and the metric predicted value are generated by processes outside the direct control of the researcher. In either case, the same mathematical description can be applied to the data (although causality is best inferred from experimental intervention).
The traditional treatment of this sort of data structure is called single-factor analysis of variance (ANOVA), or sometimes one-way ANOVA. Our Bayesian approach will be a hierarchical generalization of the traditional ANOVA model. The chapter will also consider the situation in which there is also a metric predictor that accompanies the primary nominal predictor. The metric predictor is sometimes called a covariate, and the traditional treatment of this data structure is called analysis of covariance (ANCOVA). The chapter also considers generalizations of the traditional models, because it is straight forward in Bayesian software to implement heavy-tailed distributions to accommodate outliers, along with hierarchical structure to accommodate heterogeneous variances in the different groups, etc. (Kruschke, 2015, pp. 553–554)
19.1 Describing multiple groups of metric data
Load the primary packages.
Figure 19.1 illustrates the conventional description of grouped metric data. Each group is represented as a position on the horizontal axis. The vertical axis represents the variable to be predicted by group membership. The data are assumed to be normally distributed within groups, with equal standard deviation in all groups. The group means are deflections from overall baseline, such that the deflections sum to zero. Figure 19.1 provides a specific numerical example, with data that were randomly generated from the model. (p. 554)
We’ll want a custom data-generating function for our primary group data.
generate_data <- function(seed, mean) {
set.seed(seed)
rnorm(n, mean = grand_mean + mean, sd = 2)
}
n <- 20
grand_mean <- 101
d <- tibble(group = 1:5,
deviation = c(4, -5, -2, 6, -3)) |>
mutate(d = map2(.x = group, .y = deviation, .f = generate_data)) |>
unnest(d) |>
mutate(iteration = rep(1:n, times = 5))
glimpse(d)Rows: 100
Columns: 4
$ group <int> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2,…
$ deviation <dbl> 4, 4, 4, 4, 4, 4, 4, 4, 4, 4, 4, 4, 4, 4, 4, 4, 4, 4, 4, 4, -5, -5, -5, -5, -5, -5, -5, -5, -5, -5, -5, -5, -5…
$ d <dbl> 103.74709, 105.36729, 103.32874, 108.19056, 105.65902, 103.35906, 105.97486, 106.47665, 106.15156, 104.38922, …
$ iteration <int> 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, …Here we’ll make a tibble containing the necessary data for the rotated Gaussians. As far as I can tell, Kruschke’s Gaussians only span to the bounds of percentile-based 98% intervals. We partition off those bounds for each group by the ll and ul columns in the first mutate() function. In the second mutate(), we expand the dataset to include a sequence of 100 values between those lower- and upper-limit points. In the third mutate(), we feed those points into the dnorm() function, with group-specific means and a common sd.
densities <- d |>
distinct(group, deviation) |>
mutate(ll = qnorm(0.01, mean = grand_mean + deviation, sd = 2),
ul = qnorm(0.99, mean = grand_mean + deviation, sd = 2)) |>
mutate(d = map2(.x = ll, .y = ul,
.f = \(ll, ul) seq(from = ll, to = ul, length.out = 100))) |>
mutate(density = map2(.x = d, .y = grand_mean + deviation,
.f = \(d, m) dnorm(x = d, mean = m, sd = 2))) |>
unnest(c(d, density))
head(densities)# A tibble: 6 × 6
group deviation ll ul d density
<int> <dbl> <dbl> <dbl> <dbl> <dbl>
1 1 4 100. 110. 100. 0.0133
2 1 4 100. 110. 100. 0.0148
3 1 4 100. 110. 101. 0.0165
4 1 4 100. 110. 101. 0.0183
5 1 4 100. 110. 101. 0.0203
6 1 4 100. 110. 101. 0.0224We’ll need two more supplementary tibbles to add the flourishes to the plot. The d_arrow tibble will specify our light-gray arrows. The text tibble will contain our annotation information.
d_arrow <- tibble(d = grand_mean,
group = 1:5,
deviation = c(4, -5, -2, 6, -3),
offset = 0.1)
head(d_arrow)# A tibble: 5 × 4
d group deviation offset
<dbl> <int> <dbl> <dbl>
1 101 1 4 0.1
2 101 2 -5 0.1
3 101 3 -2 0.1
4 101 4 6 0.1
5 101 5 -3 0.1d_text <- tibble(
d = grand_mean,
group = c(0:5, 0),
deviation = c(0, 4, -5, -2, 6, -3, 10),
offset = rep(c(1/4, 0), times = c(6, 1)),
angle = rep(c(90, 0), times = c(6, 1)),
label = c("beta[0]==101", "beta['[1]']==4","beta['[2]']==-5", "beta['[3]']==-2",
"beta['[4]']==6", "beta['[5]']==3", "sigma['all']==2"))
head(d_text)# A tibble: 6 × 6
d group deviation offset angle label
<dbl> <dbl> <dbl> <dbl> <dbl> <chr>
1 101 0 0 0.25 90 beta[0]==101
2 101 1 4 0.25 90 beta['[1]']==4
3 101 2 -5 0.25 90 beta['[2]']==-5
4 101 3 -2 0.25 90 beta['[3]']==-2
5 101 4 6 0.25 90 beta['[4]']==6
6 101 5 -3 0.25 90 beta['[5]']==3 We’re almost ready to plot. Before we do, let’s talk color and theme. For this chapter, we’ll take our color palette from the palettetown package (Lucas, 2016), which provides an array of color palettes inspired by Pokémon. Our color palette will be #17, which is based on Pidgeotto.
scales::show_col(pokepal(pokemon = 17))
pp <- pokepal(pokemon = 17)
pp [1] "#785848" "#E0B048" "#D03018" "#202020" "#A87858" "#F8E858" "#583828" "#E86040" "#F0F0A0" "#F8A870" "#C89878" "#F8F8F8"
[13] "#A0A0A0"Our overall plot theme will be based on the default theme_grey() with a good number of adjustments.
theme_set(
theme_grey() +
theme(text = element_text(color = pp[4]),
axis.text = element_text(color = pp[4]),
axis.ticks = element_line(color = pp[4]),
legend.background = element_blank(),
legend.box.background = element_blank(),
legend.key = element_rect(fill = pp[9]),
panel.background = element_rect(fill = pp[9], color = pp[9]),
panel.grid = element_blank(),
plot.background = element_rect(fill = pp[12], color = pp[12]),
strip.background = element_rect(fill = alpha(pp[2], 1/3), color = "transparent"),
strip.text = element_text(color = pp[4]))
)Now make Figure 19.1.
d |>
ggplot(aes(x = d, y = group, group = group)) +
geom_vline(xintercept = grand_mean, color = pp[12]) +
geom_jitter(alpha = 4/4, color = pp[10], height = 0.05, shape = 1) +
# The Gaussians
geom_ridgeline(data = densities,
aes(height = -density),
color = pp[7], fill = "transparent",
linewidth = 3/4, min_height = NA, scale = 3/2) +
# The small arrows
geom_segment(data = d_arrow,
aes(xend = d + deviation,
y = group + offset, yend = group + offset),
arrow = arrow(length = unit(0.2, "cm")),
color = pp[5], linewidth = 1) +
# The large arrow on the left
geom_segment(aes(x = 80, xend = grand_mean,
y = 0, yend = 0),
arrow = arrow(length = unit(0.2, "cm")),
color = pp[5], linewidth = 3/4) +
# The text
geom_text(data = d_text,
aes(x = grand_mean + deviation, y = group + offset,
label = label, angle = angle),
color = pp[4], parse = TRUE, size = 4) +
scale_y_continuous(NULL, breaks = 1:5,
labels = c("<1,0,0,0,0>", "<0,1,0,0,0>", "<0,0,1,0,0>", "<0,0,0,1,0>", "<0,0,0,0,1>")) +
xlab(NULL) +
coord_flip(xlim = c(90, 112),
ylim = c(-0.2, 5.5))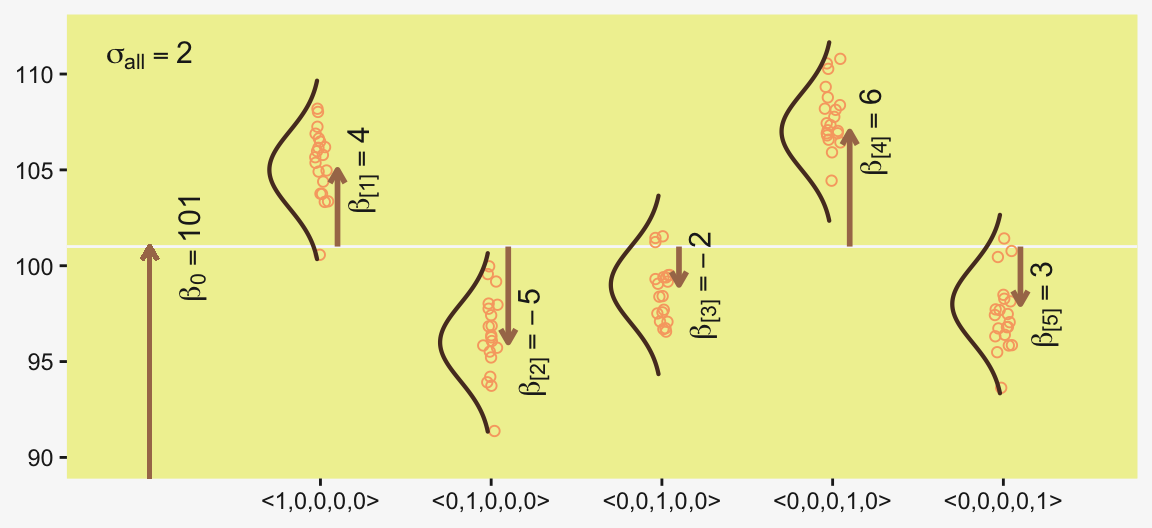
The descriptive model presented in Figure 19.1 is the traditional one used by classical ANOVA (which is described a bit more in the next section). More general models are straight forward to implement in Bayesian software. For example, outliers could be accommodated by using heavy-tailed noise distributions (such as a \(t\) distribution) instead of a normal distribution, and different groups could be given different standard deviations. (p. 556)
19.2 Traditional analysis of variance
The terminology, “analysis of variance,” comes from a decomposition of overall data variance into within-group variance and between-group variance (Fisher, 1925). Algebraically, the sum of squared deviations of the scores from their overall mean equals the sum of squared deviations of the scores from their respective group means plus the sum of squared deviations of the group means from the overall mean. In other words, the total variance can be partitioned into within-group variance plus between-group variance. Because one definition of the word “analysis” is separation into constituent parts, the term ANOVA accurately describes the underlying algebra in the traditional methods. That algebraic relation is not used in the hierarchical Bayesian approach presented here. The Bayesian method can estimate component variances, however. Therefore, the Bayesian approach is not ANOVA, but is analogous to ANOVA. (p. 556)
19.3 Hierarchical Bayesian approach
“Our goal is to estimate its parameters in a Bayesian framework. Therefore, all the parameters need to be given a meaningfully structured prior distribution” (p. 557). However, our approach will depart a little from the one in the text. All our parameters will not “have generic noncommittal prior distributions” (p. 557). Most importantly, we will not follow the example in (Gelman, 2006) of putting a broad uniform prior on \(\sigma_y\). Rather, we will continue using the half-Gaussian prior, as recommended by the Stan team. However, we will follow Kruschke’s lead for the overall intercept and use a Gaussian prior “made broad on the scale of the data” (p. 557). And like Kruschke, we will estimate \(\sigma_\beta\) from the data. To further get a sense of this, let’s make our version of the hierarchical model diagram of Figure 19.2.
# Bracket
p1 <- tibble(x = 0.99,
y = 0.5,
label = "{_}") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[8], family = "Times", hjust = 1, size = 10) +
scale_x_continuous(expand = c(0, 0), limits = c(0, 1)) +
ylim(0, 1) +
theme_void()
## Plain arrow
# Save our custom arrow settings
my_arrow <- arrow(angle = 20, length = unit(0.35, "cm"), type = "closed")
p2 <- tibble(x = 0.72,
y = 1,
xend = 0.68,
yend = 0.25) |>
ggplot(aes(x = x, xend = xend,
y = y, yend = yend)) +
geom_segment(arrow = my_arrow, color = pp[4]) +
xlim(0, 1) +
theme_void()
# Normal density
p3 <- tibble(x = seq(from = -3, to = 3, by = 0.1)) |>
ggplot(aes(x = x, y = (dnorm(x)) / max(dnorm(x)))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 0, y = 0.2,
label = "normal",
color = pp[4], size = 7) +
annotate(geom = "text",
x = c(0, 1.45), y = 0.6,
hjust = c(0.5, 0),
label = c("italic(M)[0]", "italic(S)[0]"),
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Second normal density
p4 <- tibble(x = seq(from = -3, to = 3, by = 0.1)) |>
ggplot(aes(x = x, y = (dnorm(x)) / max(dnorm(x)))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 0, y = 0.2,
label = "normal",
color = pp[4], size = 7) +
annotate(geom = "text",
x = c(0, 1.45), y = 0.6,
hjust = c(0.5, 0),
label = c("0", "sigma[beta]"),
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Two annotated arrows
p5 <- tibble(x = c(0.16, 0.81),
y = 1,
xend = c(0.47, 0.77),
yend = 0) |>
ggplot(aes(x = x, xend = xend,
y = y, yend = yend)) +
geom_segment(arrow = my_arrow, color = pp[4]) +
annotate(geom = "text",
x = c(0.25, 0.74, 0.83), y = 0.5,
label = c("'~'", "'~'", "italic(j)"),
color = pp[4], family = "Times", parse = TRUE, size = c(10, 10, 7)) +
xlim(0, 1) +
theme_void()
# Likelihood formula
p6 <- tibble(x = 0.99,
y = 0.25,
label = "beta[0]+sum()[italic(j)]*beta['['*italic(j)*']']*italic(x)['['*italic(j)*']'](italic(i))") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[4], family = "Times", hjust = 1, parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0), limits = c(0, 1)) +
ylim(0, 1) +
theme_void()
# Half-normal density
p7 <- tibble(x = seq(from = 0, to = 3, by = 0.01)) |>
ggplot(aes(x = x, y = (dnorm(x)) / max(dnorm(x)))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 1.5, y = 0.2,
label = "half-normal",
color = pp[4], size = 7) +
annotate(geom = "text",
x = 1.5, y = 0.6,
label = "0*','*~italic(S)[sigma]",
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Annotated arrow
p8 <- tibble(x = 0.38,
y = 0.65,
label = "'='") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[4], family = "Times", parse = TRUE, size = 10) +
geom_segment(x = 0.5, xend = 0.5,
y = 1, yend = 0.25,
arrow = my_arrow, color = pp[4]) +
xlim(0, 1) +
theme_void()
# The third normal density
p9 <- tibble(x = seq(from = -3, to = 3, by = 0.1)) |>
ggplot(aes(x = x, y = (dnorm(x)) / max(dnorm(x)))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 0, y = 0.2,
label = "normal",
color = pp[4], size = 7) +
annotate(geom = "text",
x = c(0, 1.45), y = 0.6,
hjust = c(0.5, 0),
label = c("mu[italic(i)]", "sigma[italic(y)]"),
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Another annotated arrow
p10 <- tibble(x = 0.55,
y = 0.6,
label = "'~'") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[4], family = "Times", parse = TRUE, size = 10) +
geom_segment(x = 0.82, xend = 0.38,
y = 1, yend = 0.2,
arrow = my_arrow, color = pp[4]) +
xlim(0, 1) +
theme_void()
# The final annotated arrow
p11 <- tibble(x = c(0.375, 0.625),
y = c(1/3, 1/3),
label = c("'~'", "italic(i)")) |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(size = c(10, 7),
color = pp[4], family = "Times", parse = TRUE) +
geom_segment(x = 0.5, xend = 0.5,
y = 1, yend = 0,
arrow = my_arrow, color = pp[4]) +
xlim(0, 1) +
theme_void()
# Some text
p12 <- tibble(x = 0.5,
y = 0.5,
label = "italic(y[i])") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[4], family = "Times", parse = TRUE, size = 7) +
xlim(0, 1) +
theme_void()
# Define the layout
layout <- c(
area(t = 1, b = 1, l = 6, r = 7),
area(t = 2, b = 3, l = 6, r = 7),
area(t = 3, b = 4, l = 1, r = 3),
area(t = 3, b = 4, l = 5, r = 7),
area(t = 6, b = 7, l = 1, r = 7),
area(t = 5, b = 6, l = 1, r = 7),
area(t = 6, b = 7, l = 9, r = 11),
area(t = 9, b = 10, l = 5, r = 7),
area(t = 8, b = 9, l = 5, r = 7),
area(t = 8, b = 9, l = 5, r = 11),
area(t = 11, b = 11, l = 5, r = 7),
area(t = 12, b = 12, l = 5, r = 7))
# Combine, adjust, and display
(p1 + p2 + p3 + p4 + p6 + p5 + p7 + p9 + p8 + p10 + p11 + p12) +
plot_layout(design = layout) &
ylim(0, 1) &
theme(plot.margin = margin(0, 5.5, 0, 5.5))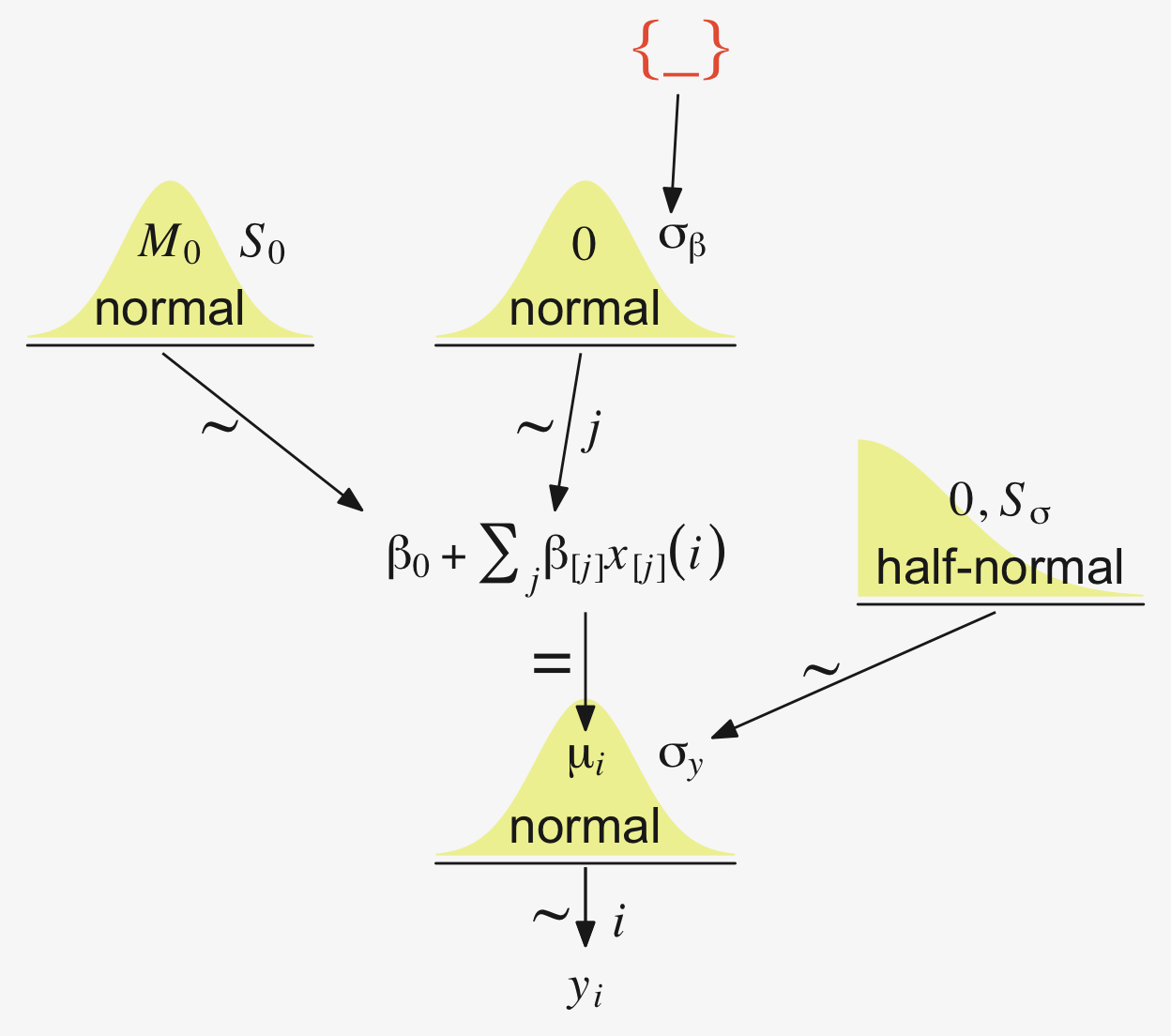
Later on in the text, Kruschke opined:
A crucial pre-requisite for estimating \(\sigma_\beta\) from all the groups is an assumption that all the groups are representative and informative for the estimate. It only makes sense to influence the estimate of one group with data from the other groups if the groups can be meaningfully described as representative of a shared higher-level distribution. (p. 559)
Although I agree with him in spirit, this doesn’t appear to strictly be the case. As odd and paradoxical as this sounds, partial pooling can be of use even when the some of the cases are of a different kind. For more on the topic, see Efron and Morris’s classic (1977) paper, Stein’s paradox in statistics, and my blog post walking out one of their examples in brms.
19.3.1 Implementation in JAGS brms
The brms setup, of course, differs a bit from JAGS.
fit <- brm(
data = my_data,
family = gaussian,
y ~ 1 + (1 | categirical_variable),
prior = c(prior(normal(0, <scale>), class = Intercept),
prior(normal(0, <scale>), class = b),
prior(cauchy(0, <scale>), class = sd),
prior(cauchy(0, <scale>), class = sigma)))The noise standard deviation \(\sigma_y\) is depicted in the prior statement including the argument class = sigma. The grand mean is depicted by the first 1 in the model formula and its prior is indicated by the class = Intercept argument. We indicate we’d like group-based deviations from the grand mean with the (1 | categirical_variable) syntax, where the 1 on the left side of the bar indicates we’d like our intercepts to vary by group and the categirical_variable part simply represents the name of a given categorical variable we’d like those intercepts to vary by. The brms default is to do this with deviance scores, the mean for which will be zero. Although it’s not obvious in the formula syntax, the model presumes the group-based deviations are normally distributed with a mean of zero and a standard deviation, which Kruschke termed \(\sigma_\beta\). There is no prior for the mean. It’s set at zero. But there is a prior for \(\sigma_\beta\), which is denoted by the argument class = sd. We, of course, are not using a uniform prior on any of our variance parameters. But in order to be weakly informative, we will use the half-Cauchy. Recall that since the brms default is to set the lower bound for any variance parameter to 0, there’s no need to worry about doing so ourselves. So even though the syntax only indicates cauchy, it’s understood to mean Cauchy with a lower bound at zero; since the mean is usually 0, that makes is a half-Cauchy.
Kruschke set the upper bound for his \(\sigma_y\) to 10 times the standard deviation of the criterion variable. The tails of the half-Cauchy are sufficiently fat that, in practice, I’ve found it doesn’t matter much what you set the \(\textit{SD}\) of its prior to. One is often a sensible default for reasonably-scaled data. But if we want to take a more principled approach, we can set it to the size of the criterion’s \(\textit{SD}\) or perhaps even 10 times that value.
Kruschke suggested using a gamma on \(\sigma_\beta\), which is a sensible alternative to half-Cauchy often used within the Stan universe. Especially in situations in which you would like to (a) keep the variance parameter above zero, but (b) still allow it to be arbitrarily close to zero, and also (c) let the likelihood dominate the posterior, the Stan team recommends the \(\operatorname{Gamma}(2, 0)\) prior, based on the paper by Chung and colleagues (2013, click here). But you should note that I don’t mean a literal 0 for the second parameter in the gamma distribution, but rather some small value like 0.1 or so. This is all clarified in Chung et al. (2013).
If you’d like that prior be even less informative, just reduce it to like \(\operatorname{Gamma}(2, 0.01)\) or so. Kruschke went further to recommend “the shape and rate parameters of the gamma distribution are set so its mode is sd(y)/2 and its standard deviation is 2*sd(y), using the function gammaShRaFromModeSD explained in Section 9.2.2.” (pp. 560–561). Let’s make that function.
gamma_a_b_from_omega_sigma <- function(mode, sd) {
if (mode <= 0) stop("`mode` must be > 0")
if (sd <= 0) stop("`sd` must be > 0")
rate <- (mode + sqrt(mode^2 + 4 * sd^2)) / (2 * sd^2)
shape <- 1 + mode * rate
list(shape = shape, rate = rate)
}So in the case of standardized data where sd(1) = 1, we’d use our gamma_a_b_from_omega_sigma() function like so.
sd_y <- 1
omega <- sd_y / 2
sigma <- 2 * sd_y
(s_r <- gamma_a_b_from_omega_sigma(mode = omega, sd = sigma))$shape
[1] 1.283196
$rate
[1] 0.5663911We might compare Kruschke’s prior to the one recommended by Chung et al in a plot.
c(prior(gamma(2, 0.1)), # Chung et al (2013)
prior(gamma(1.283196, 0.5663911))) |> # Kruschke
parse_dist() |>
ggplot(aes(y = prior, xdist = .dist_obj)) +
stat_halfeye(point_interval = mode_hdi, .width = 0.95,
color = pp[1], fill = pp[10], linewidth = 1) +
scale_x_continuous(expression(sigma[beta]~space), expand = c(0, 0)) +
scale_y_discrete(NULL, expand = expansion(mult = 0.1)) +
coord_cartesian(xlim = c(0, 65))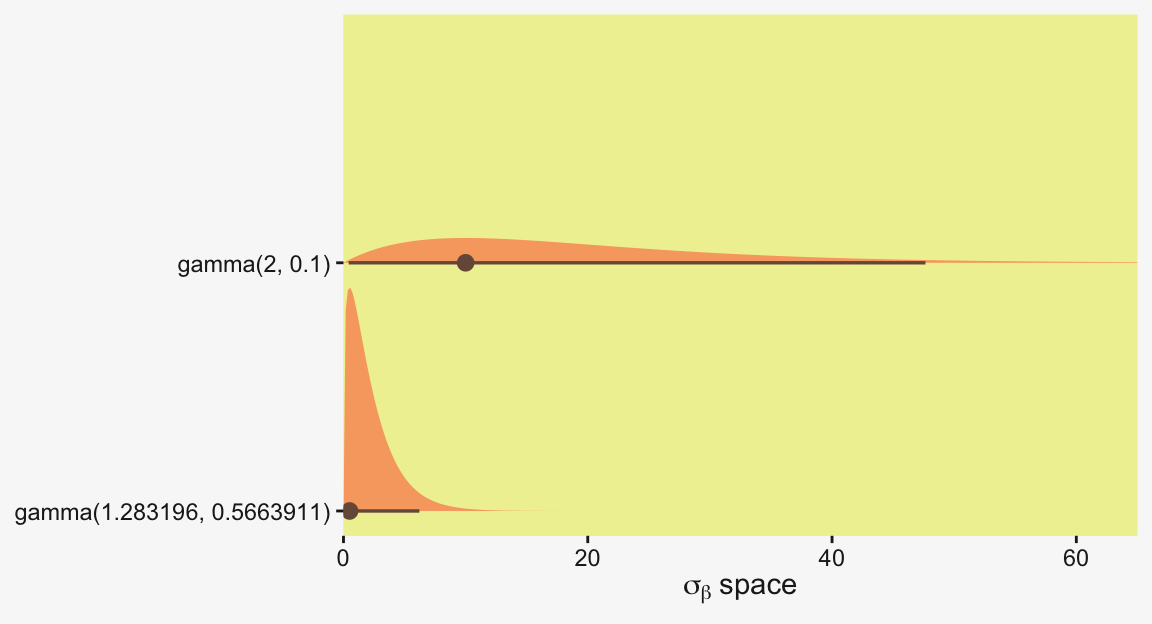
In the parameter space that matters, from zero to one, both of those gamma priors would be pretty noninformative. Computationally, both would keep the posteriors off the zero boundary, and their gentle right tails would keep the MCMC chains from spending a lot of time in ridiculous parts of the parameter space. I.e., when working with finite numbers of iterations, we want our MCMC chains wasting exactly zero iterations investigating what the density might be for \(\sigma_\beta \approx 1e10\) for standardized data.
19.3.2 Example: Sex and death
Let’s load and glimpse() at Hanley and Shapiro’s (1994) fruit-fly data.
my_data <- read_csv("data.R/FruitflyDataReduced.csv")
glimpse(my_data)Rows: 125
Columns: 3
$ Longevity <dbl> 35, 37, 49, 46, 63, 39, 46, 56, 63, 65, 56, 65, 70, 63, 65, 70, 77, 81, 86, 70, 70, 77, 77, 81, 77, 40, …
$ CompanionNumber <chr> "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", …
$ Thorax <dbl> 0.64, 0.68, 0.68, 0.72, 0.72, 0.76, 0.76, 0.76, 0.76, 0.76, 0.80, 0.80, 0.80, 0.84, 0.84, 0.84, 0.84, 0.…We can use stat_slab() to help get a sense of how our criterion Longevity is distributed across groups of CompanionNumber.
my_data |>
group_by(CompanionNumber) |>
mutate(group_mean = mean(Longevity)) |>
ungroup() |>
mutate(CompanionNumber = fct_reorder(CompanionNumber, group_mean)) |>
ggplot(aes(x = Longevity, y = CompanionNumber, fill = group_mean)) +
stat_slab(color = pp[9], linewidth = 0.2, scale = 3/2) +
scale_x_continuous(expand = expansion(mult = c(0, 0.05)), limits = c(0, NA)) +
scale_y_discrete(NULL, expand = expansion(mult = c(0, 0.4))) +
scale_fill_gradient(low = pp[4], high = pp[2], breaks = NULL) +
theme(axis.text.y = element_text(hjust = 0),
axis.ticks.y = element_blank())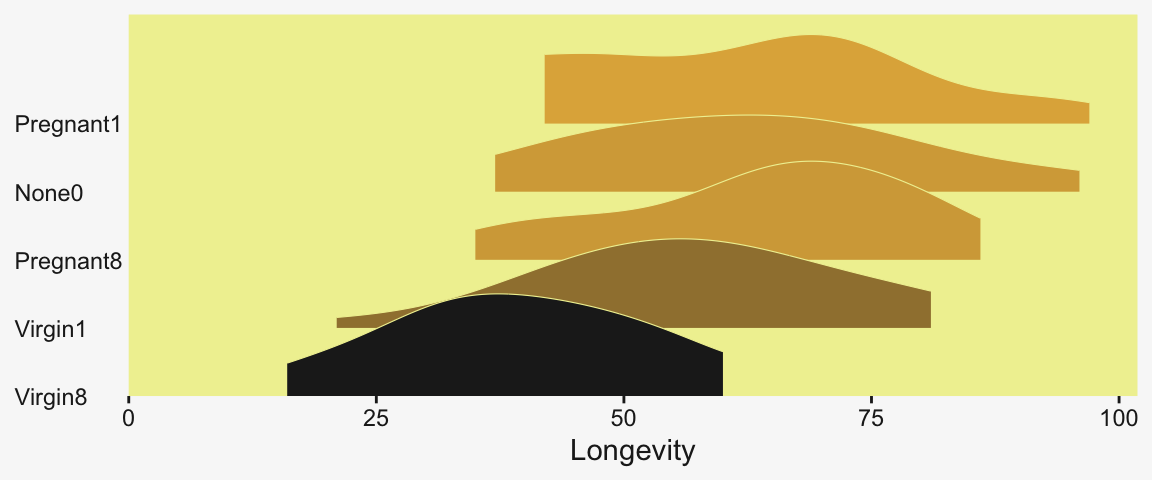
As an alternative, stat_dots() can also be fun with a plot like this.
my_data |>
group_by(CompanionNumber) |>
mutate(group_mean = mean(Longevity)) |>
ungroup() |>
mutate(CompanionNumber = fct_reorder(CompanionNumber, group_mean)) |>
ggplot(aes(x = Longevity, y = CompanionNumber, fill = group_mean)) +
stat_dots(color = pp[9], linewidth = 0.2) +
scale_x_continuous(expand = expansion(mult = c(0, 0.05)), limits = c(0, NA)) +
scale_y_discrete(NULL, expand = expansion(mult = c(0, 0.4))) +
scale_fill_gradient(low = pp[4], high = pp[2], breaks = NULL) +
theme(axis.text.y = element_text(hjust = 0),
axis.ticks.y = element_blank())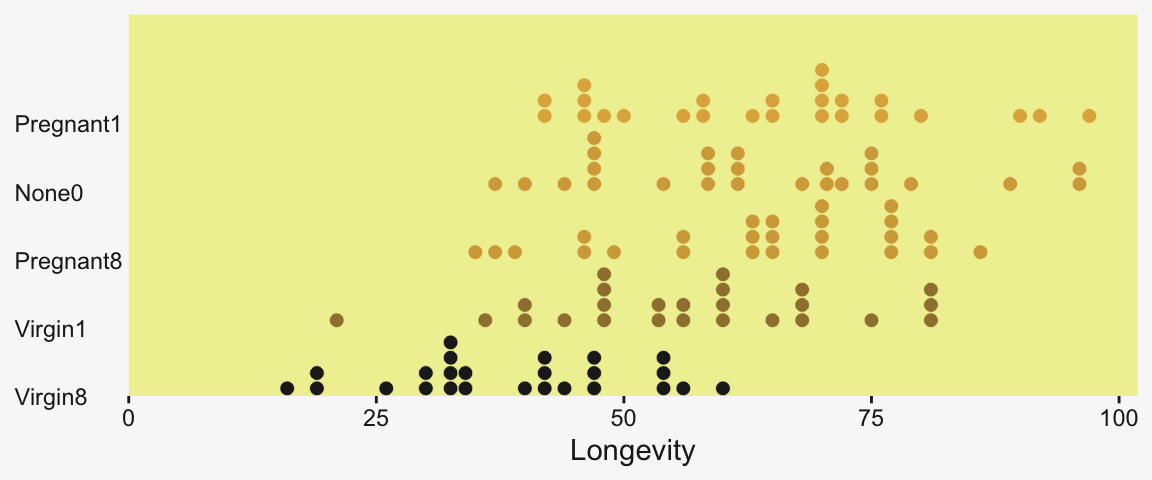
We’ll want to do the preparatory work to define our stanvars.
(mean_y <- mean(my_data$Longevity))[1] 57.44(sd_y <- sd(my_data$Longevity))[1] 17.56389omega <- sd_y / 2
sigma <- 2 * sd_y
(s_r <- gamma_a_b_from_omega_sigma(mode = omega, sd = sigma))$shape
[1] 1.283196
$rate
[1] 0.03224747With the prep work is done, here are our stanvars.
stanvars <- stanvar(mean_y, name = "mean_y") +
stanvar(sd_y, name = "sd_y") +
stanvar(s_r$shape, name = "alpha") +
stanvar(s_r$rate, name = "beta")Now fit the model, our hierarchical Bayesian alternative to an ANOVA.
fit19.1 <- brm(
data = my_data,
family = gaussian,
Longevity ~ 1 + (1 | CompanionNumber),
prior = c(prior(normal(mean_y, sd_y * 5), class = Intercept),
prior(gamma(alpha, beta), class = sd),
prior(cauchy(0, sd_y), class = sigma)),
iter = 4000, warmup = 1000, chains = 4, cores = 4,
seed = 19,
control = list(adapt_delta = 0.995),
stanvars = stanvars,
file = "fits/fit19.01")Much like Kruschke’s JAGS chains, our brms chains are well behaved, but only after fiddling with the adapt_delta setting.
color_scheme_set(scheme = pp[c(10, 8, 12, 5, 1, 4)])
plot(fit19.1, widths = c(2, 3))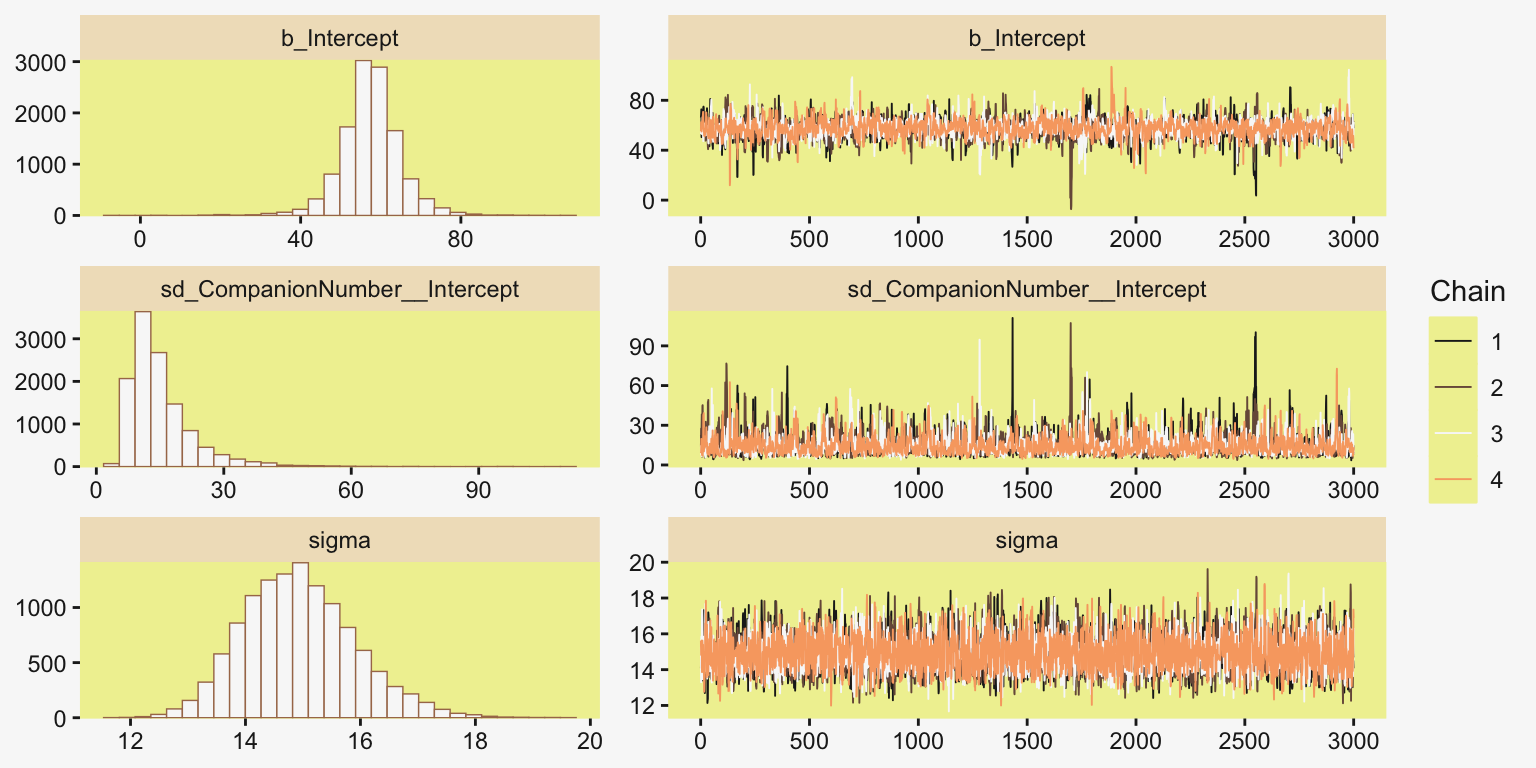
Also like Kruschke, our chains appear moderately autocorrelated.
# Extract the posterior draws
draws <- as_draws_df(fit19.1)
# Plot
draws |>
mutate(chain = .chain) |>
mcmc_acf(pars = vars(b_Intercept:sigma), lags = 10)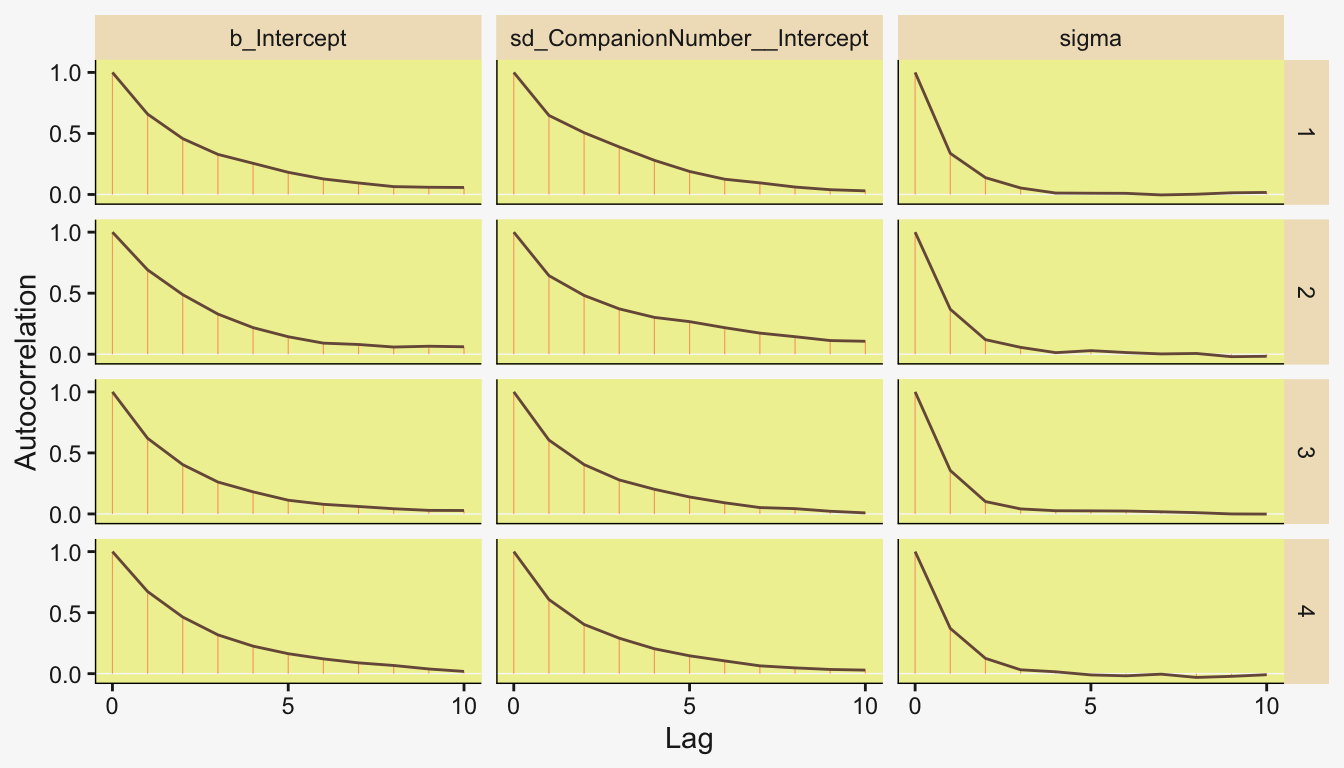
Here’s the model summary.
print(fit19.1) Family: gaussian
Links: mu = identity
Formula: Longevity ~ 1 + (1 | CompanionNumber)
Data: my_data (Number of observations: 125)
Draws: 4 chains, each with iter = 4000; warmup = 1000; thin = 1;
total post-warmup draws = 12000
Multilevel Hyperparameters:
~CompanionNumber (Number of levels: 5)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(Intercept) 15.03 7.92 6.33 35.82 1.00 2249 3252
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 57.46 7.62 42.42 72.63 1.00 2381 2824
Further Distributional Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 14.93 0.98 13.20 17.02 1.00 5792 5494
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).With the ranef() function, we can get the summaries of the group-specific deflections.
ranef(fit19.1)$CompanionNumber
, , Intercept
Estimate Est.Error Q2.5 Q97.5
None0 5.7612334 7.960761 -9.836995 22.065521
Pregnant1 6.9125893 7.965579 -8.466174 22.875082
Pregnant8 5.5780820 7.945730 -10.207332 21.718003
Virgin1 -0.6125707 7.929661 -16.261927 15.179777
Virgin8 -17.5589868 7.968122 -33.686386 -2.634376And with the coef() function, we can get those same group-level summaries in a non-deflection metric.
coef(fit19.1)$CompanionNumber
, , Intercept
Estimate Est.Error Q2.5 Q97.5
None0 63.22065 2.963166 57.46886 69.07916
Pregnant1 64.37201 2.945312 58.55072 70.15297
Pregnant8 63.03750 2.939493 57.13739 68.78137
Virgin1 56.84685 2.863651 51.27793 62.46548
Virgin8 39.90043 3.041876 33.80213 45.93965Those are all estimates of the group-specific means. Since it wasn’t modeled, all share a common \(\sigma_y\) posterior.
posterior_summary(fit19.1)["sigma", ] Estimate Est.Error Q2.5 Q97.5
14.9299370 0.9776811 13.2044010 17.0232909 To prepare for our version of the top panel of Figure 19.3, let’s make a corresponding plot of the fixed intercept, the grand mean. For this plot, we’ll use slice_sample() to randomly sample from the posterior draws.
# How many random draws from the posterior would you like?
n_draws <- 20
# Subset
set.seed(19)
draws |>
slice_sample(n = n_draws, replace = FALSE) |>
expand_grid(Longevity = 0:120) |>
mutate(density = dnorm(x = Longevity, mean = b_Intercept, sd = sigma)) |>
ggplot(aes(x = Longevity)) +
geom_line(aes(y = density, group = .draw),
alpha = 2/3, color = pp[4], linewidth = 1/3) +
geom_jitter(data = my_data,
aes(y = -0.001),
alpha = 3/4, color = pp[10], height = 0.001) +
scale_x_continuous(breaks = 0:4 * 25, limits = c(0, NA),
expand = expansion(mult = c(0, 0.05))) +
scale_y_continuous(NULL, breaks = NULL) +
labs(title = "Posterior Predictive Distribution",
subtitle = "The jittered dots are the ungrouped Longevity data. The\nGaussians are posterior draws depicting the overall\ndistribution, the grand mean.") +
coord_cartesian(xlim = c(0, 110))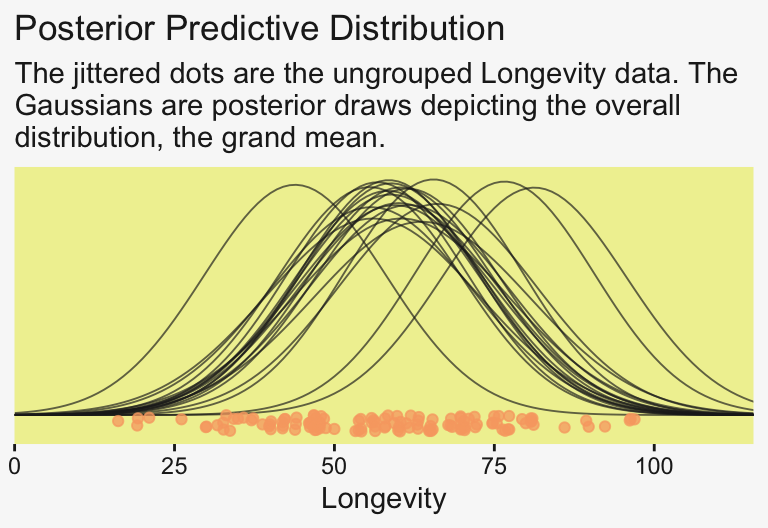
For the next plot, we’ll use a tidybayes::add_epred_draws()-based work flow instead.
densities <- my_data |>
distinct(CompanionNumber) |>
add_epred_draws(fit19.1, ndraws = n_draws, seed = 19, dpar = c("mu", "sigma"))
glimpse(densities)Rows: 100
Columns: 8
Groups: CompanionNumber, .row [5]
$ CompanionNumber <chr> "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", …
$ .row <int> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2,…
$ .chain <int> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ .iteration <int> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ .draw <int> 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11…
$ .epred <dbl> 60.98989, 63.52877, 65.20998, 63.20160, 59.04026, 59.11319, 55.11527, 65.46010, 66.03362, 63.13123, 62.1…
$ mu <dbl> 60.98989, 63.52877, 65.20998, 63.20160, 59.04026, 59.11319, 55.11527, 65.46010, 66.03362, 63.13123, 62.1…
$ sigma <dbl> 16.65716, 14.39283, 14.21772, 14.36234, 15.50326, 14.84507, 14.06252, 13.89299, 14.37961, 14.45609, 15.5…With the first two lines, we made a \(5 \times 1\) tibble containing the five levels of the experimental grouping variable, CompanionNumber. The add_epred_draws() function comes from tidybayes (see the Posterior fits section of Kay, 2021). The first argument of the add_epred_draws() is newdata, which works much like it does in brms::fitted(); it took our \(5 \times 1\) tibble. The next argument took our brms model fit, fit19.1. With the ndraws argument, we indicated we just wanted 20 random draws from the posterior. The seed argument makes those random draws reproducible. With dpar, we requested distributional regression parameters in the output. In our case, those were the \(\mu\) and \(\sigma\) values for each level of CompanionNumber. Since we took 20 draws across 5 groups, we ended up with a 100-row tibble.
The next steps are a direct extension of the method we used to make our Gaussians for our version of Figure 19.1.
densities <- densities |>
mutate(ll = qnorm(0.025, mean = mu, sd = sigma),
ul = qnorm(0.975, mean = mu, sd = sigma)) |>
mutate(Longevity = map2(.x = ll, .y = ul,
.f = \(ll, ul) seq(from = ll, to = ul, length.out = 100))) |>
unnest(Longevity) |>
mutate(density = dnorm(Longevity, mu, sigma))
glimpse(densities)Rows: 10,000
Columns: 12
Groups: CompanionNumber, .row [5]
$ CompanionNumber <chr> "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", …
$ .row <int> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1,…
$ .chain <int> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ .iteration <int> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ .draw <int> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1,…
$ .epred <dbl> 60.98989, 60.98989, 60.98989, 60.98989, 60.98989, 60.98989, 60.98989, 60.98989, 60.98989, 60.98989, 60.9…
$ mu <dbl> 60.98989, 60.98989, 60.98989, 60.98989, 60.98989, 60.98989, 60.98989, 60.98989, 60.98989, 60.98989, 60.9…
$ sigma <dbl> 16.65716, 16.65716, 16.65716, 16.65716, 16.65716, 16.65716, 16.65716, 16.65716, 16.65716, 16.65716, 16.6…
$ ll <dbl> 28.34246, 28.34246, 28.34246, 28.34246, 28.34246, 28.34246, 28.34246, 28.34246, 28.34246, 28.34246, 28.3…
$ ul <dbl> 93.63732, 93.63732, 93.63732, 93.63732, 93.63732, 93.63732, 93.63732, 93.63732, 93.63732, 93.63732, 93.6…
$ Longevity <dbl> 28.34246, 29.00201, 29.66155, 30.32110, 30.98064, 31.64018, 32.29973, 32.95927, 33.61882, 34.27836, 34.9…
$ density <dbl> 0.003508706, 0.003788874, 0.004085002, 0.004397376, 0.004726222, 0.005071701, 0.005433909, 0.005812864, …If you look at the code we used to make ll and ul, you’ll see we used 95% intervals, this time. Our second mutate() function is basically the same. After unnesting the tibble, we just needed to plug in the Longevity, mu, and sigma values into the dnorm() function to compute the corresponding density values.
densities |>
ggplot(aes(x = Longevity, y = CompanionNumber)) +
# Here we make our density lines
geom_ridgeline(aes(height = density, group = interaction(CompanionNumber, .draw)),
color = adjustcolor(pp[4], alpha.f = 2/3), fill = NA,
linewidth = 1/3, scale = 25) +
# The original data with little jitter thrown in
geom_jitter(data = my_data,
alpha = 3/4, color = pp[10], height = 0.04) +
# Pretty much everything below this line is aesthetic fluff
scale_x_continuous(breaks = 0:4 * 25, limits = c(0, 110),
expand = expansion(mult = c(0, 0.05))) +
labs(y = NULL,
title = "Data with Posterior Predictive Distrib.") +
coord_cartesian(ylim = c(1.25, 5.25)) +
theme(axis.text.y = element_text(hjust = 0),
axis.ticks.y = element_blank())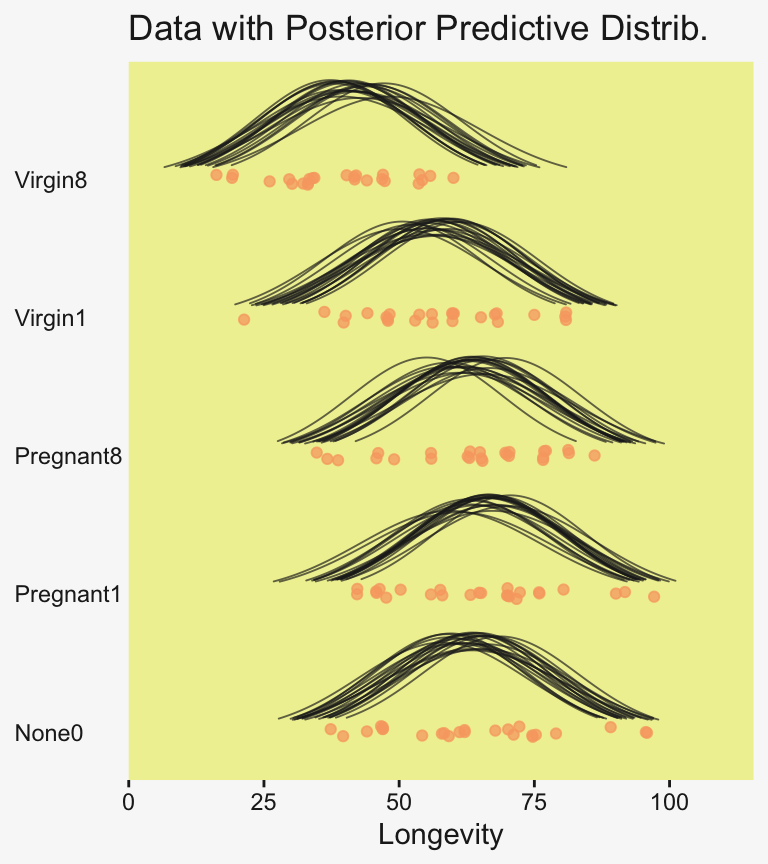
Do be aware that when you use this method, you may have to fiddle around with the geom_ridgeline() scale argument to get the Gaussian’s heights on reasonable-looking relative heights. Stick in different numbers to get a sense of what I mean. I also find that I’m often not a fan of the way the spacing on the y axis ends up with default geom_ridgeline(). It’s easy to overcome this with a little ylim fiddling.
To return to the more substantive interpretation, the top panel of
Figure 19.3 suggests that the normal distributions with homogeneous variances appear to be reasonable descriptions of the data. There are no dramatic outliers relative to the posterior predicted curves, and the spread of the data within each group appears to be reasonably matched by the width of the posterior normal curves. (Be careful when making visual assessments of homogeneity of variance because the visual spread of the data depends on the sample size; for a reminder see the [see the right panel of Figure 17.1, p. 478].) The range of credible group means, indicated by the peaks of the normal curves, suggests that the group Virgin8 is clearly lower than the others, and the group Virgin1 might be lower than the controls. To find out for sure, we need to examine the differences of group means, which we do in the next section. (p. 564)
For clarity, the “see the right panel of Figure 17.1, p. 478” part was changed following Kruschke’s Corrigenda.
19.3.3 Contrasts
It is straight forward to examine the posterior distribution of credible differences. Every step in the MCMC chain provides a combination of group means that are jointly credible, given the data. Therefore, every step in the MCMC chain provides a credible difference between groups…
To construct the credible differences of group \(1\) and group \(2\), at every step in the MCMC chain we compute
\[\begin{align*} \mu_1 - \mu_2 & = (\beta_0 + \beta_1) - (\beta_0 + \beta_2) \\ & = (+1) \cdot \beta_1 + (-1) \cdot \beta_2 \end{align*}\]
In other words, the baseline cancels out of the calculation, and the difference is a sum of weighted group deflections. Notice that the weights sum to zero. To construct the credible differences of the average of groups \(1\)-\(3\) and the average of groups \(4\)-\(5\), at every step in the MCMC chain we compute
\[\begin{align*} \small{(\mu_1 + \mu_2 + \mu_3) / 3 - (\mu_4 + \mu_5) / 2} & = \small{((\beta_0 + \beta_1) + (\beta_0 + \beta_2) + (\beta_0 + \beta_3) ) / 3 - ((\beta_0 + \beta_4) + (\beta_0 + \beta_5) ) / 2} \\ & = \small{(\beta_1 + \beta_2 + \beta_3) / 3 - (\beta_4 + \beta_5) / 2} \\ & = \small{(+ 1/3) \cdot \beta_1 + (+ 1/3) \cdot \beta_2 + (+ 1/3) \cdot \beta_3 + (- 1/2) \cdot \beta_4 + (- 1/2) \cdot \beta_5} \end{align*}\]
Again, the difference is a sum of weighted group deflections. The coefficients on the group deflections have the properties that they sum to zero, with the positive coefficients summing to \(+1\) and the negative coefficients summing to \(−1\). Such a combination is called a contrast. The differences can also be expressed in terms of effect size, by dividing the difference by \(\sigma_y\) at each step in the chain. (pp. 565–566)
To warm up, here’s how to compute the first contrast shown in the lower portion of Kruschke’s Figure 19.3–the contrast between the two pregnant conditions and the none-control condition. In this code chunk, and the code chunks to follow, I’ll be renaming the draws columns on the fly so reduce the horizontal space.
draws |>
rename(p1 = `r_CompanionNumber[Pregnant1,Intercept]`,
p8 = `r_CompanionNumber[Pregnant8,Intercept]`,
n0 = `r_CompanionNumber[None0,Intercept]`) |>
mutate(contrast = (p1 + p8) / 2 - n0) |>
ggplot(aes(x = contrast)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], slab_color = pp[5], slab_fill = pp[5]) +
scale_y_continuous(NULL, breaks = NULL) +
labs(x = expression(Difference~(mu[1]+mu[2])/2-mu[3]),
subtitle = "Pregnant1.Pregnant8 vs None0")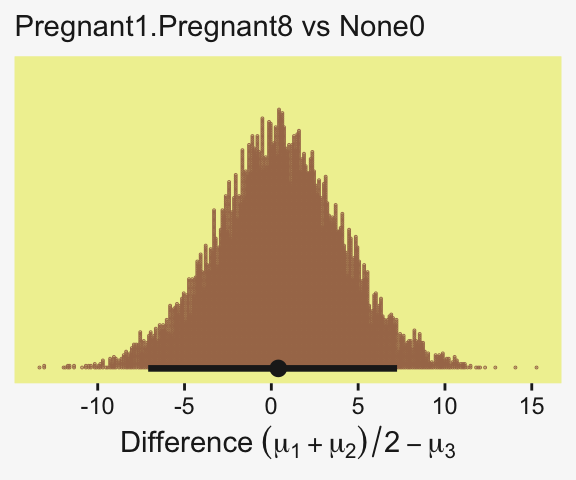
Up to this point, our primary mode of showing marginal posterior distributions has either been minute variations on Kruschke’s typical histogram approach or with densities. We’ll use those again in the future, too. In this chapter and the next, we’ll veer a little further from the source material and depict our marginal posteriors with dot plots and their very close relatives, quantile plots. In the dot plot, above, each of the 4,000 posterior draws is depicted by one of the stacked brown dots. To stack the dots in neat columns like that, tidybayes has to round a little. Though we lose something in the numeric precision, we gain a lot in interpretability. We’ll have more to say in just a moment.
In case you were curious, here are the HMC-based posterior mode and 95% HDIs.
draws |>
rename(p1 = `r_CompanionNumber[Pregnant1,Intercept]`,
p8 = `r_CompanionNumber[Pregnant8,Intercept]`,
n0 = `r_CompanionNumber[None0,Intercept]`) |>
mutate(contrast = (p1 + p8) / 2 - n0) |>
mode_hdi(contrast)# A tibble: 1 × 6
contrast .lower .upper .width .point .interval
<dbl> <dbl> <dbl> <dbl> <chr> <chr>
1 0.401 -7.09 7.24 0.95 mode hdi Little difference, there. Now let’s quantify the same contrast as a standardized mean difference (SMD) effect size.
draws |>
rename(p1 = `r_CompanionNumber[Pregnant1,Intercept]`,
p8 = `r_CompanionNumber[Pregnant8,Intercept]`,
n0 = `r_CompanionNumber[None0,Intercept]`) |>
mutate(es = ((p1 + p8) / 2 - n0) / sigma) |>
ggplot(aes(x = es)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_continuous(NULL, breaks = NULL) +
labs(x = expression(Effect~Size~(Difference/sigma[italic(y)])),
subtitle = "Pregnant1.Pregnant8 vs None0")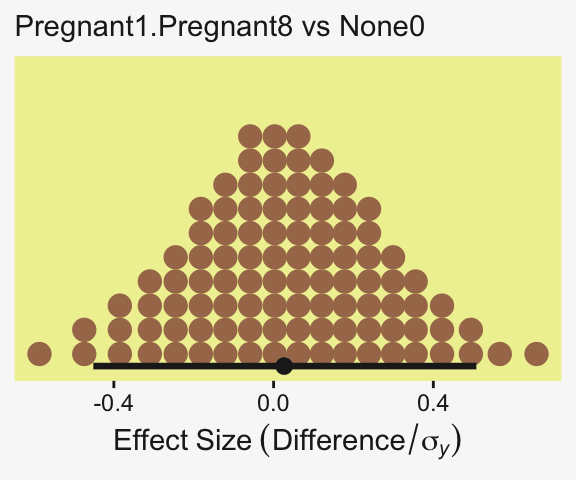
This SMD effect size is also effectively a form of a Cohen’s \(d\), which we discussed in detail in Chapter 16. In the context of this model, the \(\sigma_y\) posterior is effectively a model-based pooled standard deviation–it’s the standard deviation in the data after we have removed the variation in the group means.
We should also point out here that the non-standardized contrast we initially made above could also stand as the effect size, provided that that metric was meaningful to the target audience. But since I, for example, am not a fruit fly researcher, I find the Cohen’s \(d\) metric easier to understand. YMMV
Did you notice the quantiles = 100 argument within stat_dotsinterval()? Instead of a dot plot with 4,000 tiny little dots, that argument converted the output to a quantile plot. The 4,000 posterior draws are now summarized by 100 dots, each of which represents \(1\%\) of the total sample Fernandes et al. (2018). This quantile dot-plot method will be our main visualization approach for the rest of the chapter.
Okay, now let’s do the rest in bulk. First we’ll do the difference scores.
# Wrangle and save
differences <- draws |>
rename(n0 = `r_CompanionNumber[None0,Intercept]`,
p1 = `r_CompanionNumber[Pregnant1,Intercept]`,
p8 = `r_CompanionNumber[Pregnant8,Intercept]`,
v1 = `r_CompanionNumber[Virgin1,Intercept]`,
v8 = `r_CompanionNumber[Virgin8,Intercept]`) |>
transmute(`Pregnant1.Pregnant8.None0 vs Virgin1` = (p1 + p8 + n0) / 3 - v1,
`Virgin1 vs Virgin8` = v1 - v8,
`Pregnant1.Pregnant8.None0 vs Virgin1.Virgin8` = ((p1 + p8 + n0) / 3) - ((v1 + v8) / 2))
# Plot
differences |>
pivot_longer(cols = everything()) |>
ggplot(aes(x = value)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_continuous(NULL, breaks = NULL) +
xlab("Unstandardized Mean Difference") +
facet_wrap(~ name, scales = "free")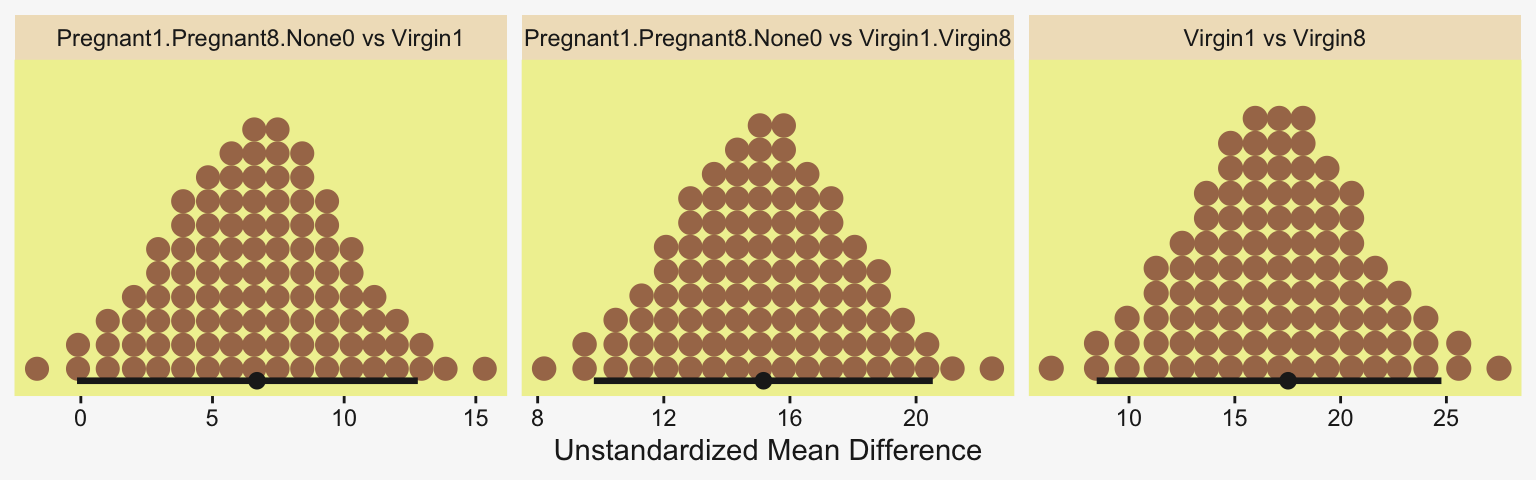
Because we save our data wrangling labor from above as differences, it won’t take much more effort to compute and plot the corresponding SMD effect sizes as displayed in the bottom row of Figure 19.3.
differences |>
mutate(across(.cols = everything(), .fns = \(x) x / draws$sigma)) |>
pivot_longer(cols = everything()) |>
ggplot(aes(x = value)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_continuous(NULL, breaks = NULL) +
xlab("Effect Size (Standardized Mean Difference)") +
facet_wrap(~ name, scales = "free_x")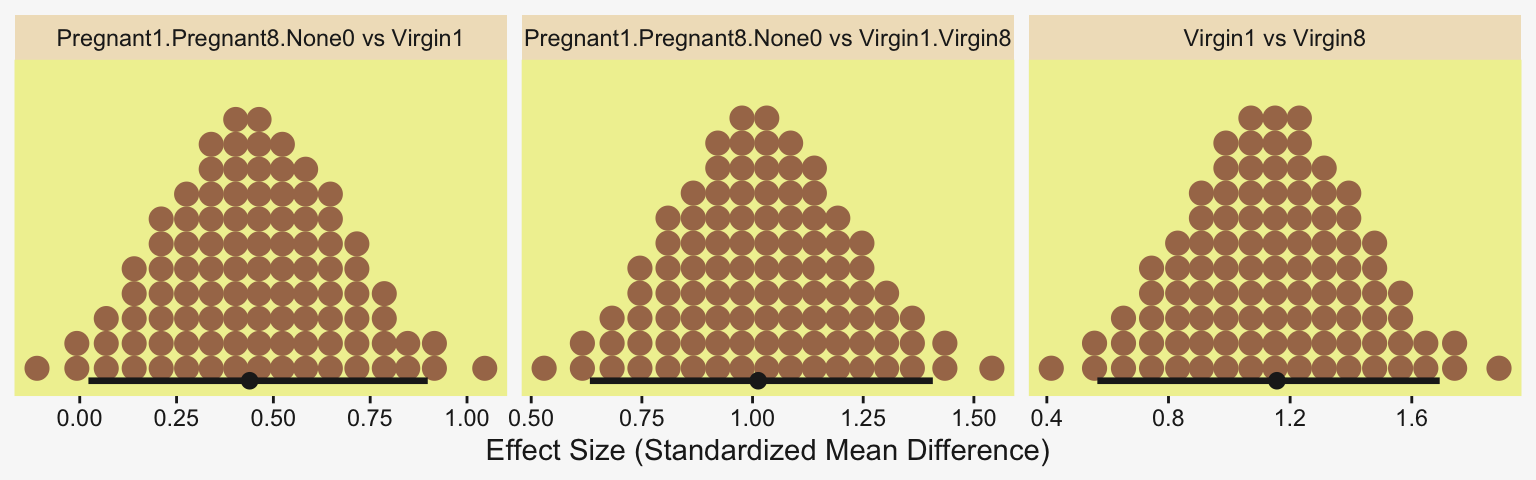
In traditional ANOVA, analysts often perform a so-called omnibus test that asks whether it is plausible that all the groups are simultaneously exactly equal. I find that the omnibus test is rarely meaningful, however…. In the hierarchical Bayesian estimation used here, there is no direct equivalent to an omnibus test in ANOVA, and the emphasis is on examining all the meaningful contrasts. (p. 567)
Speaking of all meaningful contrasts, if you’d like to make all pairwise comparisons in a hierarchical model of this form, tidybayes offers a convenient way to do so (see the Comparing levels of a factor section of Kay, 2021). Here we’ll demonstrate with stat_dotsinterval().
fit19.1 |>
# These two lines are where the magic is at
spread_draws(r_CompanionNumber[CompanionNumber, ]) |>
compare_levels(r_CompanionNumber, by = CompanionNumber) |>
ggplot(aes(x = r_CompanionNumber, y = CompanionNumber)) +
geom_vline(xintercept = 0, color = pp[12]) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
labs(x = "Contrast",
y = NULL) +
coord_cartesian(ylim = c(1.5, 10.5)) +
theme(axis.text.y = element_text(hjust = 0))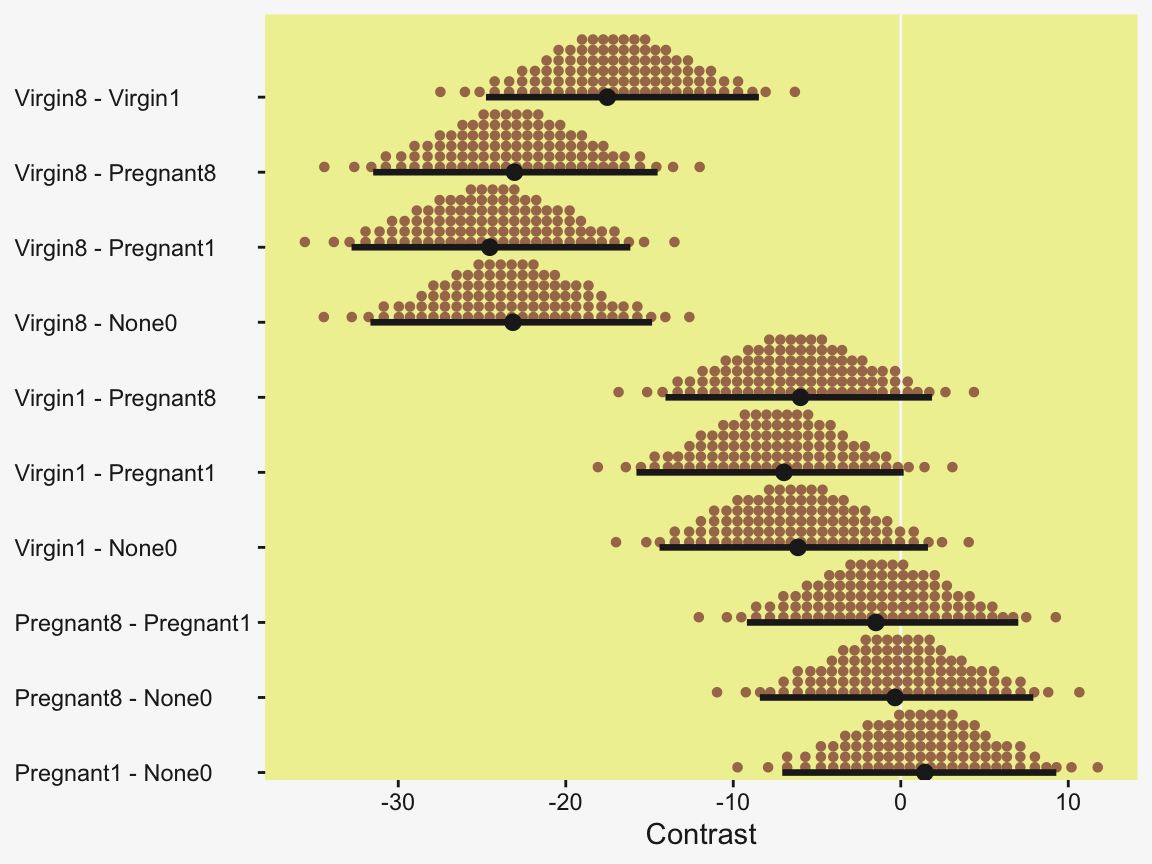
But back to that omnibus test notion. If you really wanted to, I suppose one rough analogue would be to use information criteria to compare the hierarchical model to one that includes a single intercept with no group-level deflections. Here’s what the simpler model would look like.
fit19.2 <- brm(
data = my_data,
family = gaussian,
Longevity ~ 1,
prior = c(prior(normal(mean_y, sd_y * 5), class = Intercept),
prior(cauchy(0, sd_y), class = sigma)),
iter = 4000, warmup = 1000, chains = 4, cores = 4,
seed = 19,
stanvars = stanvars,
file = "fits/fit19.02")Check the model summary.
print(fit19.2) Family: gaussian
Links: mu = identity
Formula: Longevity ~ 1
Data: my_data (Number of observations: 125)
Draws: 4 chains, each with iter = 4000; warmup = 1000; thin = 1;
total post-warmup draws = 12000
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 57.42 1.59 54.30 60.52 1.00 10897 7658
Further Distributional Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 17.67 1.12 15.63 20.02 1.00 10000 7461
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Here are their LOO values and their difference score.
fit19.1 <- add_criterion(fit19.1, criterion = "loo")
fit19.2 <- add_criterion(fit19.2, criterion = "loo")
loo_compare(fit19.1, fit19.2) |>
print(simplify = FALSE) elpd_diff se_diff elpd_loo se_elpd_loo p_loo se_p_loo looic se_looic
fit19.1 0.0 0.0 -517.7 6.9 5.5 0.6 1035.4 13.8
fit19.2 -19.3 5.3 -537.0 7.1 1.8 0.3 1074.0 14.2 The hierarchical model has a better LOO. Here are the stacking-based model weights.
(mw <- model_weights(fit19.1, fit19.2)) fit19.1 fit19.2
9.999989e-01 1.099269e-06 If you don’t like scientific notation, just round().
mw |>
round(digits = 3)fit19.1 fit19.2
1 0 Yep, in complimenting the LOO difference, virtually all the stacking weight went to the hierarchical model. You might think of this another way. The conceptual question we’re asking is: Does it make sense to say that the \(\sigma_\beta\) parameter is zero? Is zero a credible value? We’ll, I suppose we could just look at the posterior to assess for that.
draws |>
ggplot(aes(x = sd_CompanionNumber__Intercept)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_continuous(NULL, breaks = NULL) +
coord_cartesian(xlim = c(0, 50)) +
labs(x = NULL,
title = expression("Behold the fit19.1 posterior for "*sigma[beta]*"."),
subtitle = "This parameter's many things, but zero isn't one of them.")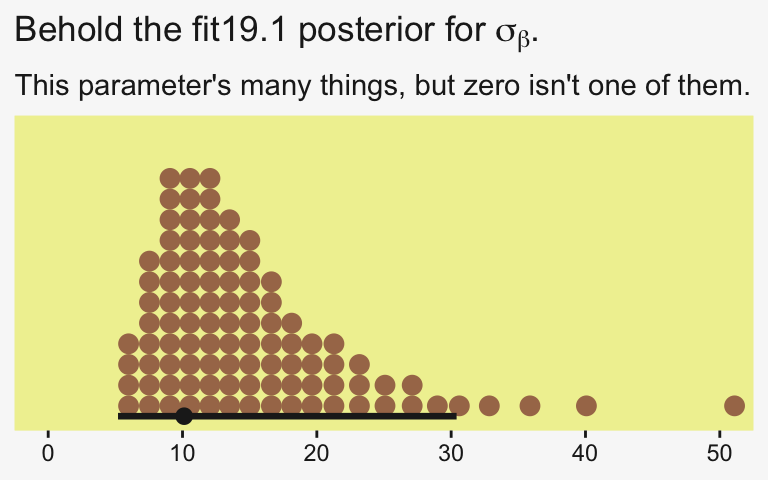
Yeah, zero and other values close to zero don’t look credible for that parameter. 95% of the mass is between 5 and 30, with the bulk hovering around 10. We don’t need an \(F\)-test or even a LOO model comparison to see the writing on wall.
19.3.4 Multiple comparisons and shrinkage
The previous section suggested that an analyst should investigate all contrasts of interest. This recommendation can be thought to conflict with traditional advice in the context on null hypothesis significance testing, which instead recommends that a minimal number of comparisons should be conducted in order to maximize the power of each test while keeping the overall false alarm rate capped at 5% (or whatever maximum is desired)…. Instead, a Bayesian analysis can mitigate false alarms by incorporating prior knowledge into the model. In particular, hierarchical structure (which is an expression of prior knowledge) produces shrinkage of estimates, and shrinkage can help rein in estimates of spurious outlying data. For example, in the posterior distribution from the fruit fly data, the modal values of the posterior group means have a range of \(23.2\). The sample means of the groups have a range of \(26.1\). Thus, there is some shrinkage in the estimated means. The amount of shrinkage is dictated only by the data and by the prior structure, not by the intended tests. (p. 568)
We may as well compute those ranges by hand. Here’s the range of the observed data.
my_data |>
group_by(CompanionNumber) |>
summarise(mean = mean(Longevity)) |>
summarise(range = max(mean) - min(mean))# A tibble: 1 × 1
range
<dbl>
1 26.1For our hierarchical model fit19.1, the posterior means are rank ordered in the same way as the empirical data.
coef(fit19.1)$CompanionNumber[, , "Intercept"] |>
data.frame() |>
rownames_to_column(var = "companion_number") |>
arrange(Estimate) |>
mutate_if(is.double, .funs = round, digits = 1) companion_number Estimate Est.Error Q2.5 Q97.5
1 Virgin8 39.9 3.0 33.8 45.9
2 Virgin1 56.8 2.9 51.3 62.5
3 Pregnant8 63.0 2.9 57.1 68.8
4 None0 63.2 3.0 57.5 69.1
5 Pregnant1 64.4 2.9 58.6 70.2If we compute the range by a difference of the point estimates of the highest and lowest posterior means, we can get a quick number.
coef(fit19.1)$CompanionNumber[, , "Intercept"] |>
as_tibble() |>
summarise(range = max(Estimate) - min(Estimate))# A tibble: 1 × 1
range
<dbl>
1 24.5Note that wasn’t fully Bayesian of us. Those means and their difference carry uncertainty and that uncertainty can be fully expressed if we use all the posterior draws (i.e., use summary = FALSE and wrangle).
coef(fit19.1, summary = FALSE)$CompanionNumber[, , "Intercept"] |>
as_tibble() |>
transmute(range = Pregnant1 - Virgin8) |>
mode_hdi(range)# A tibble: 1 × 6
range .lower .upper .width .point .interval
<dbl> <dbl> <dbl> <dbl> <chr> <chr>
1 24.5 16.1 32.8 0.95 mode hdi Happily, the central tendency of the range is near equivalent with both methods, but now we have 95% intervals, too. Do note how wide they are. This is why we work with the full set of posterior draws.
19.3.5 The two-group case
A special case of our current scenario is when there are only two groups. The model of the present section could, in principle, be applied to the two-group case, but the hierarchical structure would do little good because there is virtually no shrinkage when there are so few groups (and the top-level prior on \(\sigma_\beta\) is broad as assumed here). (p. 568)
For kicks and giggles, let’s practice. Since Pregnant1 and Virgin8 had the highest and lowest empirical means—making them the groups best suited to define our range, we’ll use them to fit the 2-group hierarchical model. To fit it with haste, just use update().
fit19.3 <- update(
fit19.1,
newdata = my_data |>
filter(CompanionNumber %in% c("Pregnant1", "Virgin8")),
seed = 19,
file = "fits/fit19.03")Even with just two groups, there were no gross issues with fitting the model.
print(fit19.3) Family: gaussian
Links: mu = identity
Formula: Longevity ~ 1 + (1 | CompanionNumber)
Data: filter(my_data, CompanionNumber %in% c("Pregnant1" (Number of observations: 50)
Draws: 4 chains, each with iter = 4000; warmup = 1000; thin = 1;
total post-warmup draws = 12000
Multilevel Hyperparameters:
~CompanionNumber (Number of levels: 2)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(Intercept) 32.95 22.22 8.95 94.60 1.00 2925 4477
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 51.11 25.09 -1.67 104.59 1.00 2830 2528
Further Distributional Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 14.22 1.49 11.64 17.48 1.00 5671 4788
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).If you compare the posteriors for \(\sigma_\beta\) across the two models, you’ll see how the one for fit19.3 is substantially larger.
posterior_summary(fit19.1)["sd_CompanionNumber__Intercept", ] Estimate Est.Error Q2.5 Q97.5
15.033545 7.921127 6.328445 35.821608 posterior_summary(fit19.3)["sd_CompanionNumber__Intercept", ] Estimate Est.Error Q2.5 Q97.5
32.946365 22.223396 8.947535 94.604415 Here that is in a coefficient plot using tidybayes::stat_interval().
bind_rows(as_draws_df(fit19.1) |> select(sd_CompanionNumber__Intercept),
as_draws_df(fit19.3) |> select(sd_CompanionNumber__Intercept)) |>
mutate(fit = rep(c("fit19.1", "fit19.3"), each = n() / 2)) |>
ggplot(aes(x = sd_CompanionNumber__Intercept, y = fit)) +
stat_interval(point_interval = mode_hdi, .width = c(0.5, 0.8, 0.95)) +
scale_color_manual(values = pp[c(11, 5, 7)],
labels = c("95%", "80%", "50%")) +
scale_x_continuous(expression(sigma[beta]),
limits = c(0, NA), expand = expansion(mult = c(0, 0.05))) +
ylab(NULL) +
theme(legend.key.size = unit(0.45, "cm"))
This all implies less shrinkage and a larger range.
coef(fit19.3, summary = FALSE)$CompanionNumber[, , "Intercept"] |>
as_tibble() |>
transmute(range = Pregnant1 - Virgin8) |>
mode_hdi(range)# A tibble: 1 × 6
range .lower .upper .width .point .interval
<dbl> <dbl> <dbl> <dbl> <chr> <chr>
1 25.5 18.1 33.8 0.95 mode hdi And indeed, the range between the two groups is larger. Now the posterior mode for their difference has almost converged to that of the raw data. Kruschke then went on to recommend using a single-level model in such situations, instead.
That is why the two-group model in Section 16.3 did not use hierarchical structure, as illustrated in Figure 16.11 (p. 468). That model also used a \(t\) distribution to accommodate outliers in the data, and that model allowed for heterogeneous variances across groups. Thus, for two groups, it is more appropriate to use the model of Section 16.3. The hierarchical multi-group model is generalized to accommodate outliers and heterogeneous variances in Section 19.5. (p. 568)
As a refresher, here’s what the brms code for that Chapter 16 model looked like.
fit16.3 <- brm(
data = my_data,
family = student,
bf(Score ~ 0 + Group,
sigma ~ 0 + Group),
prior = c(prior(normal(mean_y, sd_y * 100), class = b),
prior(normal(0, log(sd_y)), class = b, dpar = sigma),
prior(exponential(one_over_twentynine), class = nu)),
chains = 4, cores = 4,
stanvars = stanvars,
seed = 16,
file = "fits/fit16.03")Let’s adjust it for our data. Since we have a reduced data set, we’ll need to re-compute our stanvars values, which were based on the raw data.
# It's easier to just make a reduced data set
my_small_data <- my_data |>
filter(CompanionNumber %in% c("Pregnant1", "Virgin8"))
(mean_y <- mean(my_small_data$Longevity))[1] 51.76(sd_y <- sd(my_small_data$Longevity))[1] 19.11145omega <- sd_y / 2
sigma <- 2 * sd_y
(s_r <- gamma_a_b_from_omega_sigma(mode = omega, sd = sigma))$shape
[1] 1.283196
$rate
[1] 0.02963623Here we update stanvars.
stanvars <- stanvar(mean_y, name = "mean_y") +
stanvar(sd_y, name = "sd_y") +
stanvar(s_r$shape, name = "alpha") +
stanvar(s_r$rate, name = "beta") +
stanvar(1/29, name = "one_over_twentynine")Note that our priors, here, are something of a blend of those from Chapter 16 and those from our hierarchical model, fit19.1.
fit19.4 <- brm(
data = my_small_data,
family = student,
bf(Longevity ~ 0 + CompanionNumber,
sigma ~ 0 + CompanionNumber),
prior = c(prior(normal(mean_y, sd_y * 10), class = b),
prior(normal(0, log(sd_y)), class = b, dpar = sigma),
prior(exponential(one_over_twentynine), class = nu)),
iter = 4000, warmup = 1000, chains = 4, cores = 4,
seed = 19,
stanvars = stanvars,
file = "fits/fit19.04")Here’s the model summary.
print(fit19.4) Family: student
Links: mu = identity; sigma = log
Formula: Longevity ~ 0 + CompanionNumber
sigma ~ 0 + CompanionNumber
Data: my_small_data (Number of observations: 50)
Draws: 4 chains, each with iter = 4000; warmup = 1000; thin = 1;
total post-warmup draws = 12000
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
CompanionNumberPregnant1 64.63 3.27 58.00 70.96 1.00 13791 7956
CompanionNumberVirgin8 38.71 2.54 33.71 43.71 1.00 12375 7876
sigma_CompanionNumberPregnant1 2.73 0.15 2.44 3.04 1.00 12931 9309
sigma_CompanionNumberVirgin8 2.48 0.15 2.19 2.79 1.00 12608 8147
Further Distributional Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
nu 39.59 30.91 5.80 120.30 1.00 12030 8145
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Man, look at those Bulk_ESS values! As it turns out, they can be greater than the number of post-warmup samples. And here’s the range in posterior means.
fixef(fit19.4, summary = FALSE) |>
as_tibble() |>
transmute(range = CompanionNumberPregnant1 - CompanionNumberVirgin8) |>
mode_hdi(range)# A tibble: 1 × 6
range .lower .upper .width .point .interval
<dbl> <dbl> <dbl> <dbl> <chr> <chr>
1 25.8 17.7 33.8 0.95 mode hdi The results are pretty much the same as that of the two-group hierarchical model, maybe a touch larger. Yep, Kruschke was right: Hierarchical models with two groups and permissive priors on \(\sigma_\beta\) don’t shrink the estimates to the grand mean all that much.
19.4 Including a metric predictor
“In Figure 19.3, the data within each group have a large standard deviation. For example, longevity values in the Virgin8 group range from \(20\) to \(60\) days” (p. 568). Turns out Kruschke’s slightly wrong on this. Probably just a typo.
my_data |>
group_by(CompanionNumber) |>
summarise(min = min(Longevity),
max = max(Longevity),
range = max(Longevity) - min(Longevity))# A tibble: 5 × 4
CompanionNumber min max range
<chr> <dbl> <dbl> <dbl>
1 None0 37 96 59
2 Pregnant1 42 97 55
3 Pregnant8 35 86 51
4 Virgin1 21 81 60
5 Virgin8 16 60 44But you get the point. For each group, there was quite a range. We might add predictors to the model to help account for those ranges.
The additional metric predictor is sometimes called a covariate. In the experimental setting, the focus of interest is usually on the nominal predictor (i.e., the experimental treatments), and the covariate is typically thought of as an ancillary predictor to help isolate the effect of the nominal predictor. But mathematically the nominal and metric predictors have equal status in the model. Let’s denote the value of the metric covariate for subject \(i\) as \(x_\text{cov}(i)\). Then the expected value of the predicted variable for subject \(i\) is
\[\mu(i) = \beta_0 + \sum_j \beta_{[j]} x_{[j]}(i) + \beta_\text{cov} x_\text{cov}(i)\]
with the usual sum-to-zero constraint on the deflections of the nominal predictor stated in Equation 19.2. In words, Equation 19.5 says that the predicted value for subject \(i\) is a baseline plus a deflection due to the group of \(i\) plus a shift due to the value of \(i\) on the covariate. (p. 569)
And the \(j\) subscript, recall, denotes group membership. In this context, it often
makes sense to set the intercept as the mean of predicted values if the covariate is re-centered at its mean value, which is denoted \(\overline x_\text{cov}\). Therefore Equation 19.5 is algebraically reformulated to make the baseline respect those constraints…. The first equation below is simply Equation 19.5 with \(x_\text{cov}\) recentered on its mean, \(\overline x_\text{cov}\). The second line below merely algebraically rearranges the terms so that the nominal deflections sum to zero and the constants are combined into the overall baseline:
\[\begin{align*} \mu & = \alpha_0 + \sum_j \alpha_{[j]} x_{[j]} + \alpha_\text{cov} (x_\text{cov} - \overline{x}_\text{cov}) \\ & = \underbrace{\alpha_0 + \overline{\alpha} - \alpha_\text{cov} \overline{x}_\text{cov}}_{\beta_0} + \sum_j \underbrace{(\alpha_{[j]} - \overline{\alpha})}_{\beta_[j]} x_{[j]} + \underbrace{\alpha_\text{cov}}_{\beta_{\text{cov}}} x_\text{cov} \\ & \text{where } \overline{\alpha} = \frac{1}{J} \sum^J_{j = 1} \alpha_{[j]} \end{align*}\] (pp. 569–570)
We have a visual depiction of all this in the hierarchical model diagram of Figure 19.4.
# Bracket
p1 <- tibble(x = 0.99,
y = 0.5,
label = "{_}") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[8], family = "Times", hjust = 1, size = 10) +
scale_x_continuous(expand = c(0, 0), limits = c(0, 1)) +
ylim(0, 1) +
theme_void()
# Plain arrow
p2 <- tibble(x = 0.71,
y = 1,
xend = 0.68,
yend = 0.25) |>
ggplot(aes(x = x, xend = xend,
y = y, yend = yend)) +
geom_segment(arrow = my_arrow, color = pp[4]) +
xlim(0, 1) +
theme_void()
# Normal density
p3 <- tibble(x = seq(from = -3, to = 3, by = 0.1)) |>
ggplot(aes(x = x, y = (dnorm(x)) / max(dnorm(x)))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 0, y = 0.2,
label = "normal",
color = pp[4], size = 7) +
annotate(geom = "text",
x = c(0, 1.45), y = 0.6,
hjust = c(0.5, 0),
label = c("italic(M)[0]", "italic(S)[0]"),
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Second normal density
p4 <- tibble(x = seq(from = -3, to = 3, by = 0.1)) |>
ggplot(aes(x = x, y = (dnorm(x)) / max(dnorm(x)))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 0, y = 0.2,
label = "normal",
color = pp[4], size = 7) +
annotate(geom = "text",
x = c(0, 1.45), y = 0.6,
hjust = c(0.5, 0),
label = c("0", "sigma[beta]"),
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Third density
p5 <- tibble(x = seq(from = -3, to = 3, by = 0.1)) |>
ggplot(aes(x = x, y = (dnorm(x)) / max(dnorm(x)))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 0, y = 0.2,
label = "normal",
color = pp[4], size = 7) +
annotate(geom = "text",
x = c(0, 1.45), y = 0.6,
hjust = c(0.5, 0),
label = c("italic(M)[italic(c)]", "italic(S)[italic(c)]"),
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Three annotated arrows
p6 <- tibble(x = c(0.09, 0.49, 0.9),
y = 1,
xend = c(0.20, 0.40, 0.64),
yend = 0) |>
ggplot(aes(x = x, xend = xend,
y = y, yend = yend)) +
geom_segment(arrow = my_arrow, color = pp[4]) +
annotate(geom = "text",
x = c(0.11, 0.42, 0.47, 0.74), y = 0.5,
label = c("'~'", "'~'", "italic(j)", "'~'"),
size = c(10, 10, 7, 10),
color = pp[4], family = "Times", parse = TRUE) +
xlim(0, 1) +
theme_void()
# Likelihood formula
p7 <- tibble(x = 0.99,
y = 0.25,
label = "beta[0]+sum()[italic(j)]*beta['['*italic(j)*']']*italic(x)['['*italic(j)*']'](italic(i))+beta[italic(cov)]*italic(x)[italic(cov)](italic(i))") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[4], family = "Times", hjust = 1, parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0), limits = c(0, 1)) +
ylim(0, 1) +
theme_void()
# Half-normal density
p8 <- tibble(x = seq(from = 0, to = 3, by = 0.01)) |>
ggplot(aes(x = x, y = (dnorm(x)) / max(dnorm(x)))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 1.5, y = 0.2,
label = "half-normal",
color = pp[4], size = 7) +
annotate(geom = "text",
x = 1.5, y = 0.6,
label = "0*','*~italic(S)[sigma]",
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Annotated arrow
p9 <- tibble(x = 0.38,
y = 0.65,
label = "'='") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[4], family = "Times", parse = TRUE, size = 10) +
geom_segment(x = 0.5, xend = 0.5,
y = 1, yend = 0.25,
arrow = my_arrow, color = pp[4]) +
xlim(0, 1) +
theme_void()
# The fourth normal density
p10 <- tibble(x = seq(from = -3, to = 3, by = 0.1)) |>
ggplot(aes(x = x, y = (dnorm(x)) / max(dnorm(x)))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 0, y = 0.2,
label = "normal",
color = pp[4], size = 7) +
annotate(geom = "text",
x = c(0, 1.45), y = 0.6,
hjust = c(0.5, 0),
label = c("mu[italic(i)]", "sigma[italic(y)]"),
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Another annotated arrow
p11 <- tibble(x = 0.5,
y = 0.6,
label = "'~'") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[4], family = "Times", parse = TRUE, size = 10) +
geom_segment(x = 0.85, xend = 0.27,
y = 1, yend = 0.2,
arrow = my_arrow, color = pp[4]) +
xlim(0, 1) +
theme_void()
# The final annotated arrow
p12 <- tibble(x = c(0.375, 0.625),
y = c(1/3, 1/3),
label = c("'~'", "italic(i)")) |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(size = c(10, 7),
color = pp[4], family = "Times", parse = TRUE) +
geom_segment(x = 0.5, xend = 0.5,
y = 1, yend = 0,
arrow = my_arrow, color = pp[4]) +
xlim(0, 1) +
theme_void()
# Some text
p13 <- tibble(x = 0.5,
y = 0.5,
label = "italic(y[i])") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[4], family = "Times", parse = TRUE, size = 7) +
xlim(0, 1) +
theme_void()
# Define the layout
layout <- c(
area(t = 1, b = 1, l = 6, r = 7),
area(t = 2, b = 3, l = 6, r = 7),
area(t = 3, b = 4, l = 1, r = 3),
area(t = 3, b = 4, l = 5, r = 7),
area(t = 3, b = 4, l = 9, r = 11),
area(t = 6, b = 7, l = 1, r = 9),
area(t = 5, b = 6, l = 1, r = 11),
area(t = 6, b = 7, l = 11, r = 13),
area(t = 9, b = 10, l = 5, r = 7),
area(t = 8, b = 9, l = 5, r = 7),
area(t = 8, b = 9, l = 5, r = 13),
area(t = 11, b = 11, l = 5, r = 7),
area(t = 12, b = 12, l = 5, r = 7))
# Combine, adjust, and display
(p1 + p2 + p3 + p4 + p5 + p7 + p6 + p8 + p10 + p9 + p11 + p12 + p13) +
plot_layout(design = layout) &
ylim(0, 1) &
theme(plot.margin = margin(0, 5.5, 0, 5.5))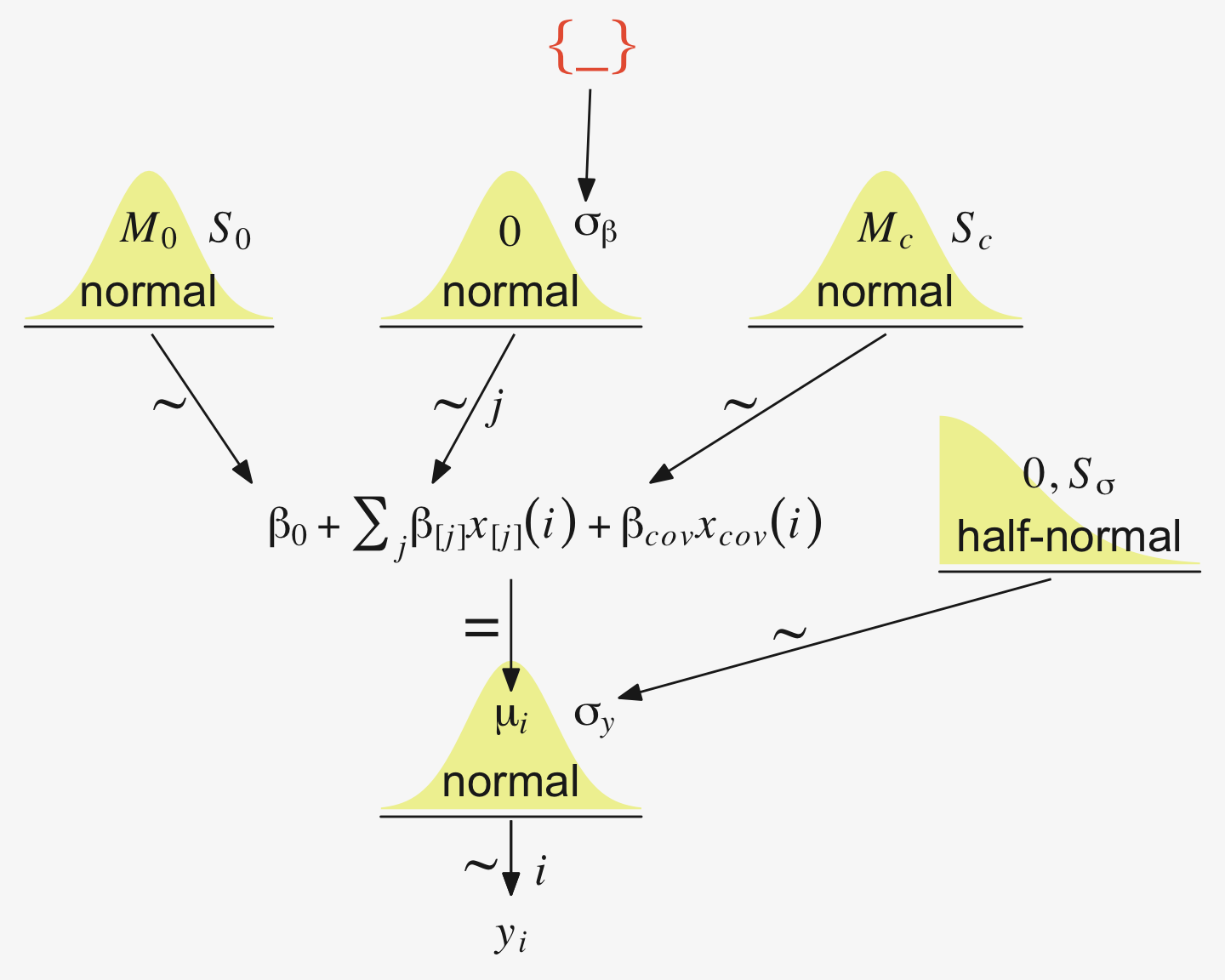
19.4.1 Example: Sex, death, and size
Kruschke recalled fit19.1’s estimate for \(\sigma_y\) had a posterior mode around 14.8. Let’s confirm with a plot.
as_draws_df(fit19.1) |>
ggplot(aes(x = sigma)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_continuous(NULL, breaks = NULL) +
xlab(expression(sigma[italic(y)])) +
theme(panel.grid = element_blank())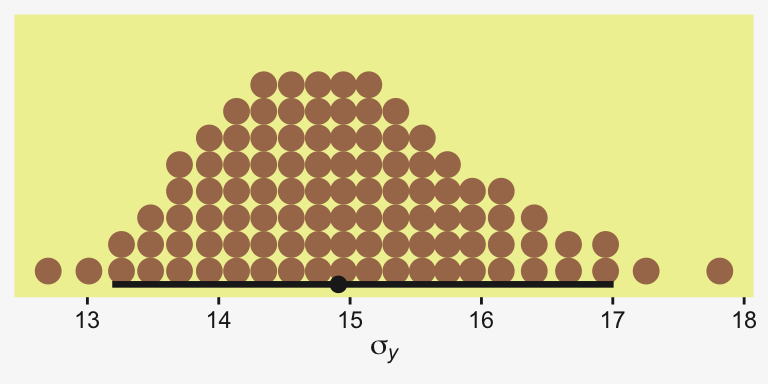
Yep, that looks about right. That large of a difference in days would indeed make it difficult to detect between-group differences if those differences were typically on the scale of just a few days. Since Thorax is moderately correlated with Longevity, including Thorax in the statistical model should help shrink that \(\sigma_y\) estimate, making it easier to compare group means. Following the sensibilities from the equations just above, here we’ll mean-center our covariate, first.
my_data <- my_data |>
mutate(thorax_c = Thorax - mean(Thorax))
head(my_data)# A tibble: 6 × 4
Longevity CompanionNumber Thorax thorax_c
<dbl> <chr> <dbl> <dbl>
1 35 Pregnant8 0.64 -0.181
2 37 Pregnant8 0.68 -0.141
3 49 Pregnant8 0.68 -0.141
4 46 Pregnant8 0.72 -0.101
5 63 Pregnant8 0.72 -0.101
6 39 Pregnant8 0.76 -0.0610Our model code follows the structure of that in Kruschke’s Jags-Ymet-Xnom1met1-MnormalHom-Example.R and Jags-Ymet-Xnom1met1-MnormalHom.R files. As a preparatory step, we redefine the values necessary for stanvars.
(mean_y <- mean(my_data$Longevity))[1] 57.44(sd_y <- sd(my_data$Longevity))[1] 17.56389(sd_thorax_c <- sd(my_data$thorax_c))[1] 0.07745367omega <- sd_y / 2
sigma <- 2 * sd_y
(s_r <- gamma_a_b_from_omega_sigma(mode = omega, sd = sigma))$shape
[1] 1.283196
$rate
[1] 0.03224747stanvars <- stanvar(mean_y, name = "mean_y") +
stanvar(sd_y, name = "sd_y") +
stanvar(sd_thorax_c, name = "sd_thorax_c") +
stanvar(s_r$shape, name = "alpha") +
stanvar(s_r$rate, name = "beta")Now we’re ready to fit the brm() model, our hierarchical alternative to ANCOVA.
fit19.5 <- brm(
data = my_data,
family = gaussian,
Longevity ~ 1 + thorax_c + (1 | CompanionNumber),
prior = c(prior(normal(mean_y, sd_y * 5), class = Intercept),
prior(normal(0, 2 * sd_y / sd_thorax_c), class = b),
prior(gamma(alpha, beta), class = sd),
prior(cauchy(0, sd_y), class = sigma)),
iter = 4000, warmup = 1000, chains = 4, cores = 4,
seed = 19,
control = list(adapt_delta = 0.99),
stanvars = stanvars,
file = "fits/fit19.05")Here’s the model summary.
print(fit19.5) Family: gaussian
Links: mu = identity
Formula: Longevity ~ 1 + thorax_c + (1 | CompanionNumber)
Data: my_data (Number of observations: 125)
Draws: 4 chains, each with iter = 4000; warmup = 1000; thin = 1;
total post-warmup draws = 12000
Multilevel Hyperparameters:
~CompanionNumber (Number of levels: 5)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(Intercept) 13.88 7.45 6.02 33.61 1.00 2552 3470
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 57.72 7.21 43.64 71.68 1.00 2549 3045
thorax_c 135.86 12.40 111.34 160.56 1.00 6904 6947
Further Distributional Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 10.60 0.69 9.34 12.06 1.00 7607 6752
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Let’s see if that \(\sigma_y\) posterior shrank.
as_draws_df(fit19.5) |>
ggplot(aes(x = sigma)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_continuous(NULL, breaks = NULL) +
xlab(expression(sigma[italic(y)]))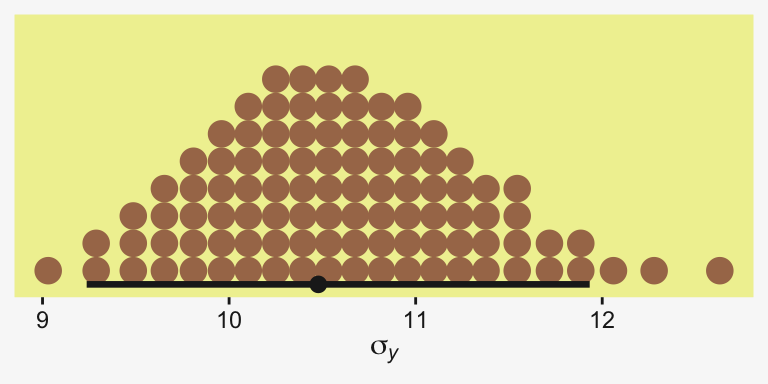
Yep, sure did! Now our between-group comparisons should be more precise. Heck, if we wanted to we could even make a difference plot for the \(\sigma_y\) posteriors.
tibble(sigma1 = as_draws_df(fit19.1) |> pull(sigma),
sigma5 = as_draws_df(fit19.5) |> pull(sigma)) |>
transmute(dif = sigma1 - sigma5) |>
ggplot(aes(x = dif)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_continuous(NULL, breaks = NULL) +
labs(x = expression(sigma[italic(y)][" | fit19.1"]-sigma[italic(y)][" | fit19.5"]),
title = "This is a difference distribution")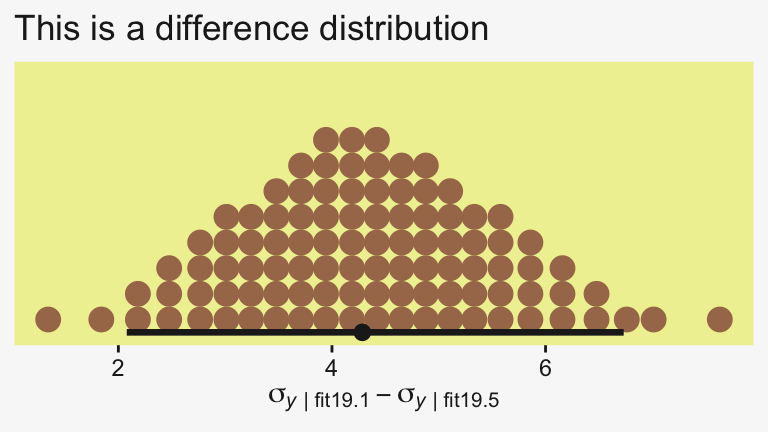
If you want a quick and dirty plot of the relation between thorax_c and Longevity, you might employ brms::conditional_effects().
conditional_effects(fit19.5) |>
plot(line_args = list(color = pp[5], fill = pp[11]))Ignoring unknown labels:
• fill : "NA"
• colour : "NA"
Ignoring unknown labels:
• fill : "NA"
• colour : "NA"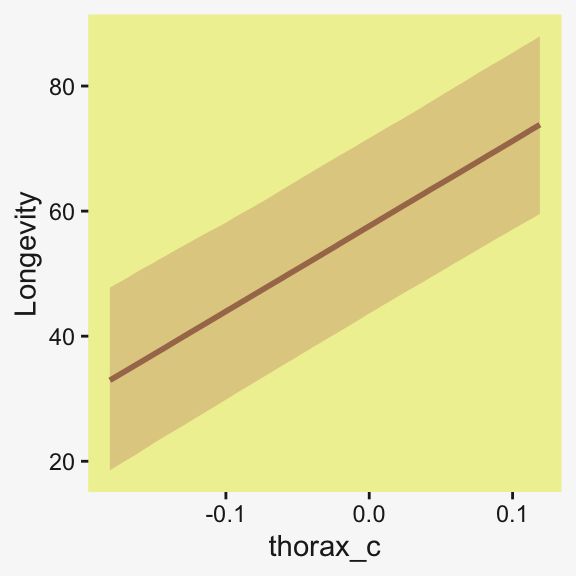
But to make plots like the ones at the top of Figure 19.5, we’ll have to work a little harder. First, we need some intermediary values marking off the three values along the Thorax-axis Kruschke singled out in his top panel plots. As far as I can tell, they were the min(), the max(), and their mean().
(r <- range(my_data$Thorax))[1] 0.64 0.94mean(r)[1] 0.79Next, we’ll make the data necessary for our side-tipped Gaussians. For kicks and giggles, we’ll choose 80 draws instead of 20. But do note how we used our r values, from above, to specify both Thorax and thorax_c values in addition to the CompanionNumber categories for the newdata argument. Otherwise, this workflow is very much the same as in previous plots.
n_draws <- 80
densities <- my_data |>
distinct(CompanionNumber) |>
expand_grid(Thorax = c(r[1], mean(r), r[2])) |>
mutate(thorax_c = Thorax - mean(my_data$Thorax)) |>
add_epred_draws(fit19.5, ndraws = n_draws, seed = 19, dpar = c("mu", "sigma")) |>
mutate(ll = qnorm(0.025, mean = mu, sd = sigma),
ul = qnorm(0.975, mean = mu, sd = sigma)) |>
mutate(Longevity = map2(.x = ll, .y = ul, .f = \(ll, ul) seq(from = ll, to = ul, length.out = 100))) |>
unnest(Longevity) |>
mutate(density = dnorm(x = Longevity, mean = mu, sd = sigma))
glimpse(densities)Rows: 120,000
Columns: 14
Groups: CompanionNumber, Thorax, thorax_c, .row [15]
$ CompanionNumber <chr> "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", …
$ Thorax <dbl> 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.…
$ thorax_c <dbl> -0.18096, -0.18096, -0.18096, -0.18096, -0.18096, -0.18096, -0.18096, -0.18096, -0.18096, -0.18096, -0.1…
$ .row <int> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1,…
$ .chain <int> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ .iteration <int> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ .draw <int> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1,…
$ .epred <dbl> 39.17164, 39.17164, 39.17164, 39.17164, 39.17164, 39.17164, 39.17164, 39.17164, 39.17164, 39.17164, 39.1…
$ mu <dbl> 39.17164, 39.17164, 39.17164, 39.17164, 39.17164, 39.17164, 39.17164, 39.17164, 39.17164, 39.17164, 39.1…
$ sigma <dbl> 11.60219, 11.60219, 11.60219, 11.60219, 11.60219, 11.60219, 11.60219, 11.60219, 11.60219, 11.60219, 11.6…
$ ll <dbl> 16.43175, 16.43175, 16.43175, 16.43175, 16.43175, 16.43175, 16.43175, 16.43175, 16.43175, 16.43175, 16.4…
$ ul <dbl> 61.91152, 61.91152, 61.91152, 61.91152, 61.91152, 61.91152, 61.91152, 61.91152, 61.91152, 61.91152, 61.9…
$ Longevity <dbl> 16.43175, 16.89114, 17.35054, 17.80993, 18.26932, 18.72871, 19.18810, 19.64749, 20.10689, 20.56628, 21.0…
$ density <dbl> 0.005037415, 0.005439648, 0.005864798, 0.006313270, 0.006785390, 0.007281391, 0.007801409, 0.008345471, …Here, we’ll use a simplified workflow to extract the fitted() values in order to make the regression lines. Since these are straight lines, all we need are two values for each draw, one at the extremes of the Thorax axis.
f <- my_data |>
distinct(CompanionNumber) |>
expand_grid(Thorax = c(r[1], mean(r), r[2])) |>
mutate(thorax_c = Thorax - mean(my_data$Thorax)) |>
add_epred_draws(fit19.5, ndraws = n_draws, seed = 19, value = "Longevity")
glimpse(f)Rows: 1,200
Columns: 8
Groups: CompanionNumber, Thorax, thorax_c, .row [15]
$ CompanionNumber <chr> "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", "Pregnant8", …
$ Thorax <dbl> 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.64, 0.…
$ thorax_c <dbl> -0.18096, -0.18096, -0.18096, -0.18096, -0.18096, -0.18096, -0.18096, -0.18096, -0.18096, -0.18096, -0.1…
$ .row <int> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1,…
$ .chain <int> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ .iteration <int> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ .draw <int> 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 2…
$ Longevity <dbl> 39.17164, 38.72720, 41.58792, 39.72092, 44.90579, 34.93792, 44.02521, 43.36934, 42.00286, 42.27404, 38.6…Now we’re ready to make our plots for the top row of Figure 19.3.
densities |>
ggplot(aes(x = Longevity, y = Thorax)) +
# The Gaussians
geom_ridgeline(aes(height = -density, group = interaction(Thorax, .draw)),
color = adjustcolor(pp[4], alpha.f = 1/5), fill = NA,
linewidth = 1/5, min_height = NA, scale = 5/3) +
# The vertical lines below the Gaussians
geom_line(aes(group = interaction(Thorax, .draw)),
alpha = 1/5, color = pp[4], linewidth = 1/5) +
# The regression lines
geom_line(data = f,
aes(group = .draw),
alpha = 1/5, color = pp[4], linewidth = 1/5) +
# The data
geom_point(data = my_data,
alpha = 3/4, color = pp[10]) +
coord_flip(xlim = c(0, 110),
ylim = c(0.58, 1)) +
facet_wrap(~ CompanionNumber, ncol = 5)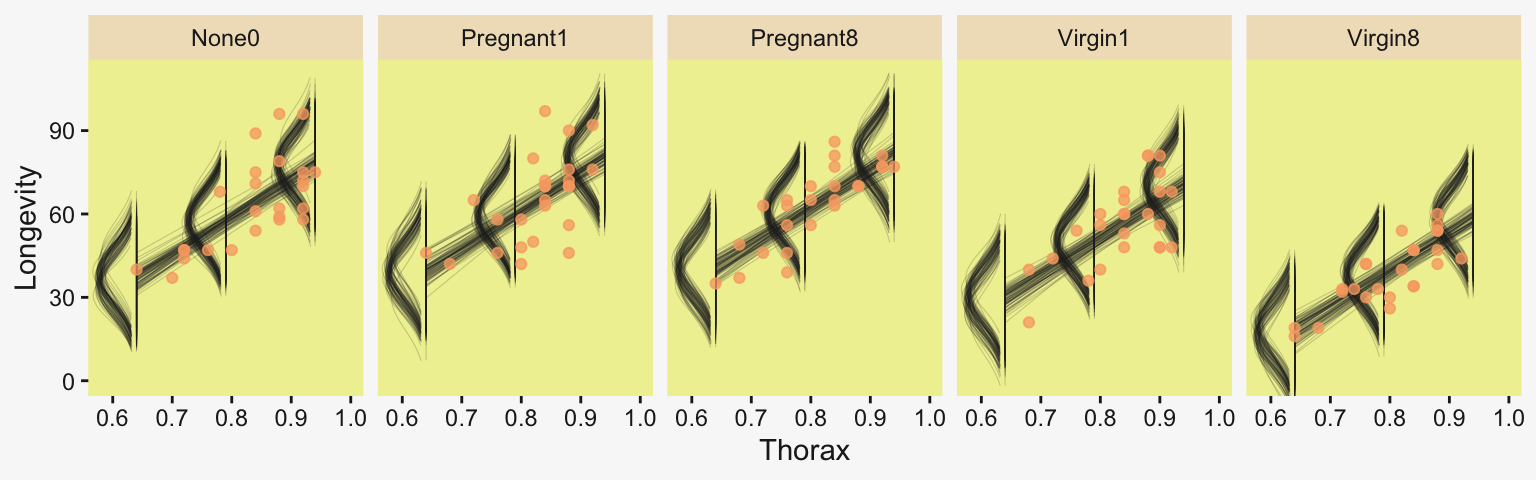
Now we have a covariate in the model, we have to decide on which of its values we want to base our group comparisons. Unless there’s a substantive reason for another value, the mean is a good standard choice. And since the covariate thorax_c is already mean centered, that means we can effectively leave it out of the equation. Here we make and save them in the simple difference metric.
draws <- as_draws_df(fit19.5)
differences <- draws |>
rename(n0 = `r_CompanionNumber[None0,Intercept]`,
p1 = `r_CompanionNumber[Pregnant1,Intercept]`,
p8 = `r_CompanionNumber[Pregnant8,Intercept]`,
v1 = `r_CompanionNumber[Virgin1,Intercept]`,
v8 = `r_CompanionNumber[Virgin8,Intercept]`) |>
transmute(`Pregnant1.Pregnant8 vs None0` = (p1 + p8) / 2 - n0,
`Pregnant1.Pregnant8.None0 vs Virgin1` = (p1 + p8 + n0) / 3 - v1,
`Virgin1 vs Virgin8` = v1 - v8,
`Pregnant1.Pregnant8.None0 vs Virgin1.Virgin8` = ((p1 + p8 + n0) / 3) - ((v1 + v8) / 2))
p1 <- differences |>
pivot_longer(cols = everything()) |>
ggplot(aes(x = value)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_continuous(NULL, breaks = NULL) +
xlab("Difference") +
theme(strip.text = element_text(size = 6.4)) +
facet_wrap(~ name, ncol = 4, scales = "free_x")Now we’ll look at the differences in the effect size metric. Since we saved our leg work above, it’s really easy to just convert the differences in bulk with mutate_all(). After the conversion, we’ll bind the two rows of subplots together with a little patchwork and display the results.
p2 <- differences |>
mutate(across(.cols = everything(), .fns = \(x) x / draws$sigma)) |>
pivot_longer(cols = everything()) |>
ggplot(aes(x = value)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_continuous(NULL, breaks = NULL) +
xlab("Effect size (SMD)") +
theme(strip.text = element_text(size = 6.4)) +
facet_wrap(~ name, ncol = 4, scales = "free_x")
# Combine
p1 / p2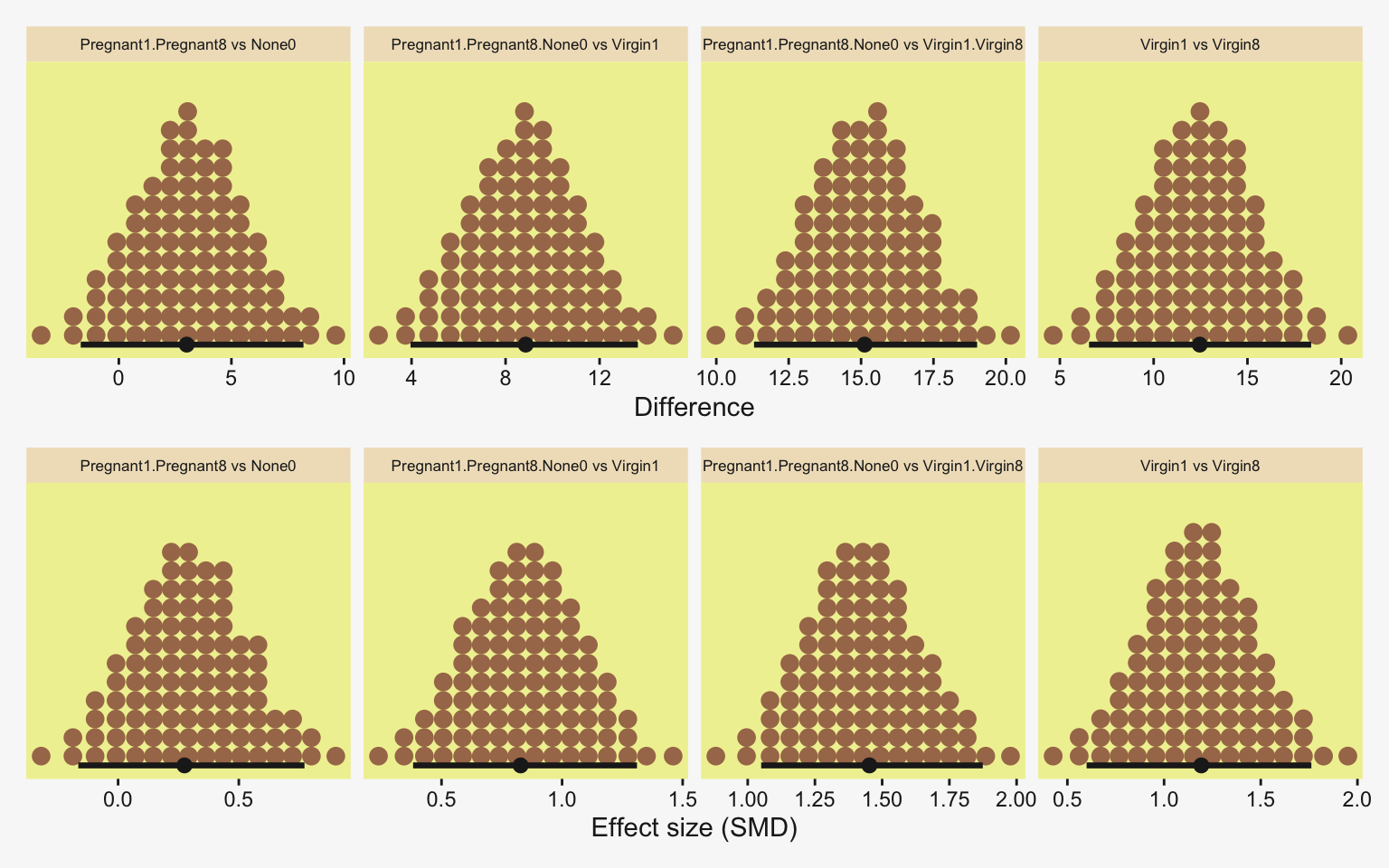
“The HDI widths of all the contrasts have gotten smaller by virtue of including the covariate in the analysis” (p. 571).
19.4.1.1 Bonus: What was that standardizer?
When we compute an SMD effect size, the typical standardizer is the pooled standard deviation. The pooled standard deviation, recall, is the standard deviation of the criterion variable, after you have removed the variation from the group variable. Here’s how we might compute the pooled standard deviation from the sample data.
sd_p <- my_data |>
group_by(CompanionNumber) |>
summarise(s = sd(Longevity)) |>
summarise(sqrt(sum(s^2) / n())) |>
pull()
# What?
sd_p[1] 14.80808Much like we learned in Chapter 16, the \(\sigma_y\) posterior in a Bayesian Gaussian ANOVA is effectively a population estimate for the pooled standard deviation. This holds for multilevel ANOVAs. However, this is not the case for Bayesian Gaussian ANCOVAs, multilevel or not. For the Gaussian ANCOVA, \(\sigma_y\) is now the posterior for the residual standard deviation, which has both the group-level variation, and the covariance with the covariate[s] removed. To get a sense, we make a plot of the \(\sigma_y\) posteriors for our multilevel ANOVA and ANCOVA, and compare them with our sd_p value above.
bind_rows(as_draws_df(fit19.1) |> select(sigma),
as_draws_df(fit19.5) |> select(sigma)) |>
mutate(fit = rep(c("fit19.1 (ANOVA)", "fit19.5 (ANCOVA)"), each = n() / 2)) |>
ggplot(aes(x = sigma, y = fit)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
geom_vline(xintercept = sd_p, color = pp[13], linetype = 2) +
scale_y_discrete(NULL, expand = expansion(mult = 0.1)) +
labs(x = expression(sigma[italic(y)]),
title = expression('For the Gaussian ANCOVA, '*italic(SD[p])!=sigma[italic(y)]),
subtitle = expression("The dashed line marks the sample statistic "*italic(s[p])*"."))
To the extent the covariate[s] in the ANCOVA covary with the criterion, \(\sigma_{y_\text{ANCOVA}} < \sigma_{y_\text{ANOVA}}\). This systemic difference will have consequences for using \(\sigma_{y_\text{ANCOVA}}\) to standardize the SMD effect sizes. Another issue is one of interpretation. The pooled standard deviation is the conventional standardizer, which means your scientific colleagues will likely have been socialized into thinking in those terms. I’m not sure how to even interpret a SMD standardized with a pooled residual standard deviation. What would such a thing mean? To give a sense of the difference in values from using \(\sigma_{y_\text{ANCOVA}}\), here we compare those SMD posteriors with alternative posteriors standardized with the sample pooled standard deviation, \(s_p\).
differences |>
mutate(sigma_y = draws$sigma) |>
pivot_longer(cols = -sigma_y, values_to = "contrast") |>
mutate(`sigma[italic(y)]` = contrast / sigma_y,
`italic(s[p])` = contrast / sd_p) |>
pivot_longer(cols = contains("["), names_to = "standardizer") |>
ggplot(aes(x = value, y = standardizer)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_discrete(expand = expansion(mult = 0.04), labels = ggplot2:::parse_safe) +
xlab("Effect size (SMD, by standardizer)") +
theme(strip.text = element_text(size = 5.7)) +
facet_wrap(~ name, ncol = 4, scales = "free_x")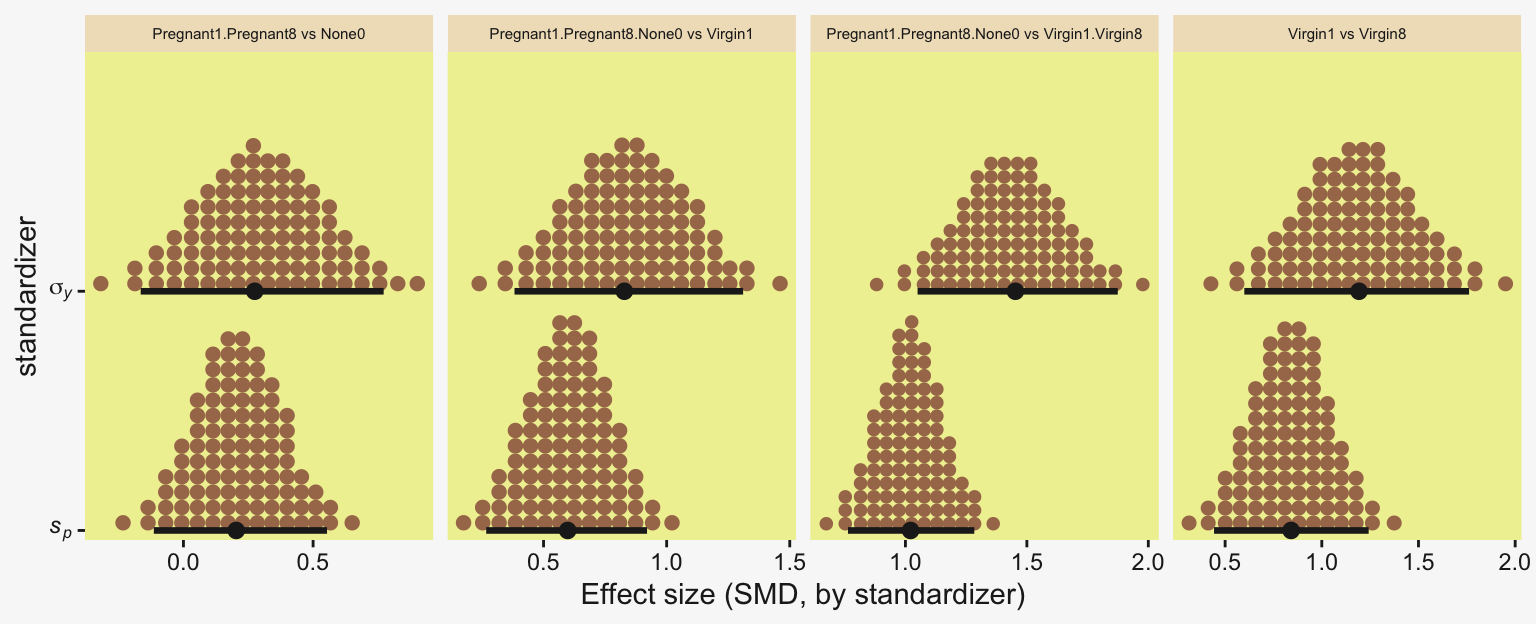
Because \(\sigma_{y_\text{ANCOVA}}\) is systemically smaller than \(s_p\), the SMD posteriors are systemically larger when you use it as the standardizer. But larger isn’t always better, because in this context we may have lost interpretability. Without further justification from Kruschke or others, I would recommend against using \(\sigma_{y_\text{ANCOVA}}\), and instead recommend using either the sample estimate for the pooled standard deviation, \(s_p\), or any well-established population value if one is available in a given area (which doesn’t seem to be the case here).
19.4.2 Analogous to traditional ANCOVA
In contrast with ANCOVA,
Bayesian methods do not partition the least-squares variance to make estimates, and therefore the Bayesian method is analogous to ANCOVA but is not ANCOVA. Frequentist practitioners are urged to test (with \(p\) values) whether the assumptions of (a) equal slope in all groups, (b) equal standard deviation in all groups, and (c) normally distributed noise can be rejected. In a Bayesian approach, the descriptive model is generalized to address these concerns, as will be discussed in Section 19.5. (p. 572)
19.4.3 Relation to hierarchical linear regression
Here Kruschke contrasts our last model with the one from way back in Section 17.3. As a refresher, here’s what that code looked like.
fit17.4 <- brm(
data = my_data,
family = student,
y_z ~ 1 + x_z + (1 + x_z || Subj),
prior = c(prior(normal(0, 10), class = Intercept),
prior(normal(0, 10), class = b),
prior(normal(0, 1), class = sigma),
# The next line is new
prior(normal(0, 1), class = sd),
prior(exponential(one_over_twentynine) + 1, class = nu)),
chains = 4, cores = 4,
stanvars = stanvar(1/29, name = "one_over_twentynine"),
seed = 17,
file = "fits/fit17.04")And for convenience, here’s the code from the model we just fit.
fit19.5 <- brm(
data = my_data,
family = gaussian,
Longevity ~ 1 + thorax_c + (1 | CompanionNumber),
prior = c(prior(normal(mean_y, sd_y * 5), class = Intercept),
prior(normal(0, 2 * sd_y / sd_thorax_c), class = b),
prior(gamma(alpha, beta), class = sd),
prior(cauchy(0, sd_y), class = sigma)),
iter = 4000, warmup = 1000, chains = 4, cores = 4,
seed = 19,
control = list(adapt_delta = 0.99),
stanvars = stanvars,
file = "fits/fit19.05")It’s easy to get lost in the differences in the priors and the technical details with the model chains and such. The main thing to notice, here, is the differences in the model formulas (i.e., the likelihoods). Both models had intercepts and slopes. But whereas the model from Section 17.3 set both parameters to random, only the intercept in our last model was random. The covariate thorax_c was fixed–it did not vary by group. Had we wanted it to, our formula syntax would have been something like Longevity ~ 1 + thorax_c + (1 + thorax_c || CompanionNumber). And again, as noted in Section 17.3.1, the || portion of the syntax set the random intercepts and slopes to be orthogonal (i.e., correlate exactly at zero). As we’ll see, this will often not be the case. But let’s not get ahead of ourselves.
Conceptually, the main difference between the models is merely the focus of attention. In the hierarchical linear regression model, the focus was on the slope coefficient. In that case, we were trying to estimate the magnitude of the slope, simultaneously for individuals and overall. The intercepts, which describe the levels of the nominal predictor, were of ancillary interest. In the present section, on the other hand, the focus of attention is reversed. We are most interested in the intercepts and their differences between groups, with the slopes on the covariate being of ancillary interest. (p. 573)
19.4.4 Bonus: ANHECOVA sideshow
Though our focus here was on the “intercepts and their differences between groups,” it’s not unreasonable to consider adding random slopes. Thinking back to the single-level case, this could look like a model formula of
brm(
y ~ 1 + group + x + group:x,
...)where the final group:x term allows the slope for the covariate x to vary across each levels of group. In the context of a multilevel model, this might look like
brm(
y ~ 1 + x + (1 + x | group),
...)where, in principle, you could also leave out the correlation between random intercepts and slopes with the || syntax. In some parts of the literature, the single-level version of this model has been called the analysis of heterogeneous covariance [ANHECOVA; e.g., Ye et al. (2022)]. I’m not aware it has been extended much to the multilevel context, but here we’ll give it a shot.
fit19.5b <- brm(
data = my_data,
family = gaussian,
Longevity ~ 1 + thorax_c + (1 + thorax_c | CompanionNumber),
prior = c(prior(normal(mean_y, sd_y * 5), class = Intercept),
prior(normal(0, 2 * sd_y / sd_thorax_c), class = b),
prior(gamma(alpha, beta), class = sd),
prior(cauchy(0, sd_y), class = sigma),
prior(lkj(2), class = cor)),
iter = 4000, warmup = 1000, chains = 4, cores = 4,
seed = 19,
control = list(adapt_delta = 0.99),
stanvars = stanvars,
file = "fits/fit19.05b")Did this model capture important differences in thorax_c slopes? We might check with a faceted plot.
xmin <- min(my_data$thorax_c)
xmax <- max(my_data$thorax_c)
nd <- my_data |>
distinct(CompanionNumber) |>
expand_grid(thorax_c = seq(from = xmin, to = xmax, length.out = 30))
nd |>
add_fitted_draws(fit19.5b) |>
rename(Longevity = .value) |>
ggplot(aes(x = thorax_c, y = Longevity)) +
stat_lineribbon(point_interval = mode_hdi, .width = 0.95,
color = pp[4], fill = pp[11], linewidth = 1/2) +
geom_point(data = my_data,
color = pp[10]) +
facet_wrap(~ CompanionNumber, nrow = 1)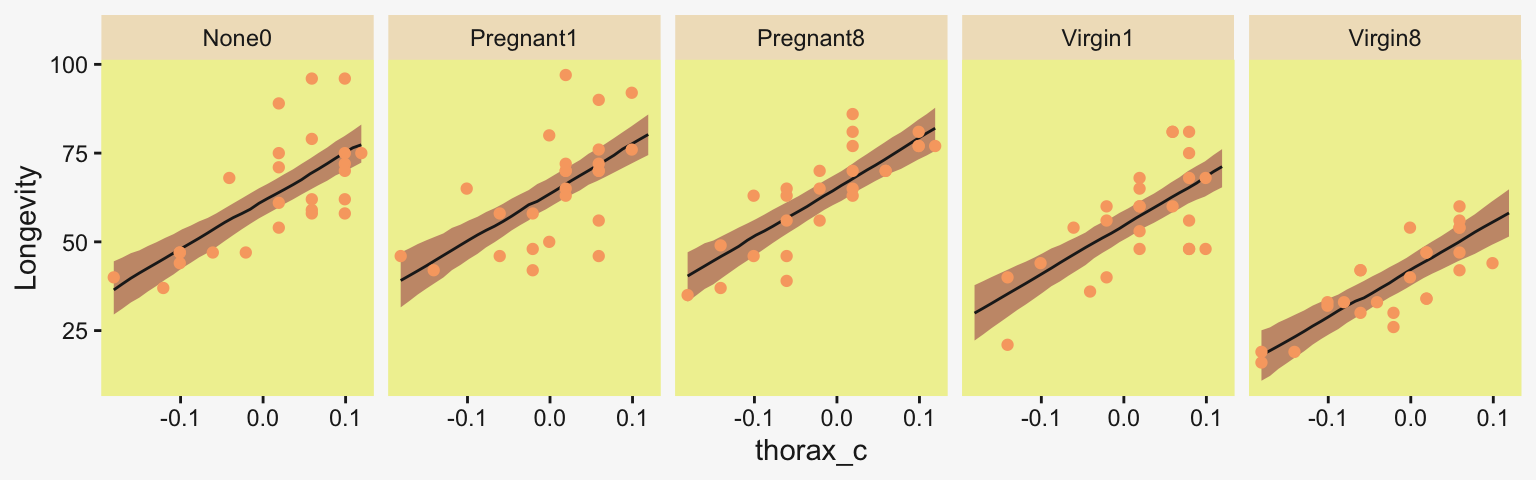
In this case, no, the covariate slopes were not dramatically different across the level of CompanionNumber. In my experience, this is often the case. But in principle it might not always be so. Who knows? Your next use case might one of those exceptions for which this matters.
Keep in mind, though, that the slopes themselves are still a sideshow for the ANHECOVA. The SMD effect sizes are still the main event. When you use a mean-centered covariate (as we did with thorax_c), you can compute the group contrasts for the ANHECOVA in the same way you did with the ANCOVA. Here we compute then, and compare them with the ANCOVA contrasts in a coefficient plot.
# Compute and save the contrasts for the multilevel ANHECOVA
differences_anhecova <- as_draws_df(fit19.5b) |>
rename(n0 = `r_CompanionNumber[None0,Intercept]`,
p1 = `r_CompanionNumber[Pregnant1,Intercept]`,
p8 = `r_CompanionNumber[Pregnant8,Intercept]`,
v1 = `r_CompanionNumber[Virgin1,Intercept]`,
v8 = `r_CompanionNumber[Virgin8,Intercept]`) |>
transmute(`Pregnant1.Pregnant8 vs None0` = (p1 + p8) / 2 - n0,
`Pregnant1.Pregnant8.None0 vs Virgin1` = (p1 + p8 + n0) / 3 - v1,
`Virgin1 vs Virgin8` = v1 - v8,
`Pregnant1.Pregnant8.None0 vs Virgin1.Virgin8` = ((p1 + p8 + n0) / 3) - ((v1 + v8) / 2))
# Wrangle and plot
differences |>
bind_rows(differences_anhecova) |>
mutate(model = rep(c("ANCOVA", "ANHECOVA"), each = n() / 2)) |>
pivot_longer(cols = -model) |>
mutate(value = value / sd_p) |>
ggplot(aes(x = value, y = model)) +
stat_pointinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4]) +
labs(x = expression("SMD effect size (standardized by "*italic(s[p])*")"),
y = NULL) +
theme(strip.text = element_text(size = 6)) +
facet_wrap(~ name, nrow = 1)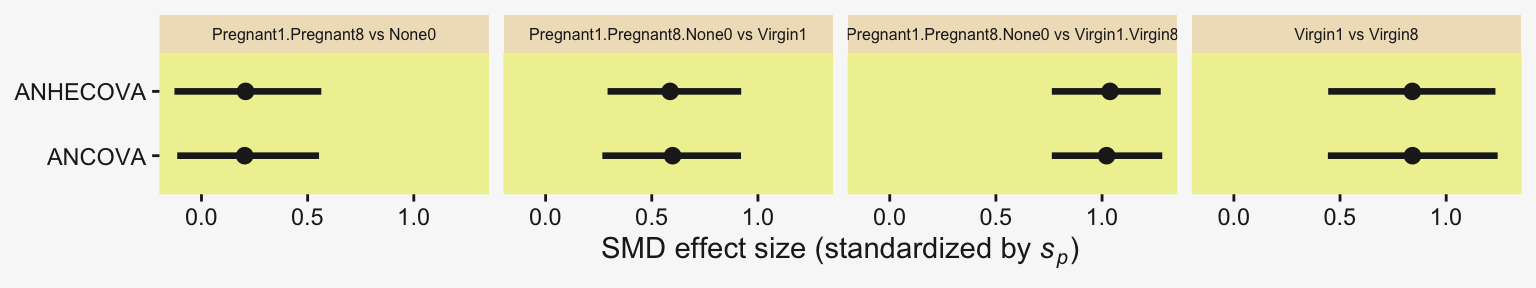
The ANHECOVA contrasts were about the same as those from the ANCOVA, which has also generally been my experience.
19.5 Heterogeneous variances and robustness against outliers
In Kruschke’s opening paragraph, we read:
We can relax those assumptions in Bayesian software. In this section, we use \(t\) distributed noise instead of normal distributions, and we provide every group with its own standard-deviation parameter. Moreover, we put a hierarchical prior on the standard-deviation parameters, so that each group mutually informs the standard deviations of the other groups via the higher-level distribution. (p. 573)
This language is misleading. The Student-\(t\) distribution does not have a “standard-deviation parameter.” Rather, it has scale and normality parameters \(\sigma\) and \(\nu\), and provided \(\nu > 2\), its standard deviation is a function of both following the form \(\sigma \sqrt{\nu / (\nu - 2)}\). We covered this issue extensively in Section 16.5, but it has become relevant again in this chapter. As we will see as we go along, this will also have consequences for how we compute effect sizes.
In Figure 19.6 on page 574, Kruschke laid out the diagram for a hierarchical Student’s-\(t\) model in for which both the \(\mu\) and \(\sigma\) parameters are random. If you recall, Bürkner (2022) calls these distributional models and they are indeed available within the brms framework. But there’s a catch. Though we can model \(\sigma\) all day long and we can even make it hierarchical, brms limits us to modeling the hierarchical \(\sigma\) parameters within the typical GLMM framework. That is, we will depart from Kruschke’s schematic in that we will be
- modeling the log of \(\sigma\),
- indicating its grand mean with the
sigma ~ 1syntax, - modeling the group-level deflections as Gaussian with a mean of 0 and standard deviation \(\sigma_\sigma\) estimated from the data, and
- choosing a sensible prior for \(\sigma_\sigma\) that is left-bound at 0 and gently slopes to the right (i.e., a folded \(t\) or gamma distribution).
Thus, here’s our brms-centric variant of the diagram in Figure 19.6.
# Bracket
p1 <- tibble(x = 0.99,
y = 0.5,
label = "{_}") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[8], family = "Times", hjust = 1, size = 10) +
scale_x_continuous(expand = c(0, 0), limits = c(0, 1)) +
ylim(0, 1) +
theme_void()
# Plain arrow
p2 <- tibble(x = 0.68,
y = 1,
xend = 0.68,
yend = 0.25) |>
ggplot(aes(x = x, xend = xend,
y = y, yend = yend)) +
geom_segment(arrow = my_arrow, color = pp[4]) +
xlim(0, 1) +
theme_void()
# Normal density
p3 <- tibble(x = seq(from = -3, to = 3, by = 0.1)) |>
ggplot(aes(x = x, y = (dnorm(x)) / max(dnorm(x)))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 0, y = 0.2,
label = "normal",
color = pp[4], size = 7) +
annotate(geom = "text",
x = c(0, 1.45), y = 0.6,
hjust = c(0.5, 0),
label = c("italic(M)[0]", "italic(S)[0]"),
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Second normal density
p4 <- tibble(x = seq(from = -3, to = 3, by = 0.1)) |>
ggplot(aes(x = x, y = (dnorm(x)) / max(dnorm(x)))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 0, y = 0.2,
label = "normal",
color = pp[4], size = 7) +
annotate(geom = "text",
x = c(0, 1.45), y = 0.6,
hjust = c(0.5, 0),
label = c("0", "sigma[beta]"),
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Third normal density
p5 <- tibble(x = seq(from = -3, to = 3, by = 0.1)) |>
ggplot(aes(x = x, y = (dnorm(x)) / max(dnorm(x)))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 0, y = 0.2,
label = "normal",
color = pp[4], size = 7) +
annotate(geom = "text",
x = c(0, 1.15), y = 0.6,
label = c("italic(M)[mu[sigma]]", "italic(S)[mu[sigma]]"),
hjust = c(0.5, 0),
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Half-normal density
p6 <- tibble(x = seq(from = 0, to = 3, by = 0.01)) |>
ggplot(aes(x = x, y = (dnorm(x)) / max(dnorm(x)))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 1.5, y = 0.2,
label = "half-normal",
size = 6, color = pp[4]) +
annotate(geom = "text",
x = 1.5, y = 0.6,
label = "0*','*~italic(S)[sigma[sigma]]",
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Exponential density
p7 <- tibble(x = seq(from = 0, to = 1, by = 0.01)) |>
ggplot(aes(x = x, y = (dexp(x, 2) / max(dexp(x, 2))))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 0.5, y = 0.2,
label = "exp",
color = pp[4], size = 7) +
annotate(geom = "text",
x = 0.5, y = 0.6,
label = "italic(K)",
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Likelihood formula
p8 <- tibble(x = 0.5,
y = 0.25,
label = "beta[0]+sum()[italic(j)]*beta['['*italic(j)*']']*italic(x)['['*italic(j)*']'](italic(i))") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0), limits = c(0, 1)) +
ylim(0, 1) +
theme_void()
# Normal density
p9 <- tibble(x = seq(from = -3, to = 3, by = 0.1)) |>
ggplot(aes(x = x, y = (dnorm(x)) / max(dnorm(x)))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 0, y = 0.2,
label = "normal",
color = pp[4], size = 7) +
annotate(geom = "text",
x = c(0, 1.2), y = 0.6,
hjust = c(0.5, 0),
label = c("mu[sigma]", "sigma[sigma]"),
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Four annotated arrows
p10 <- tibble(x = c(0.06, 0.37, 0.67, 0.95),
y = 1,
xend = c(0.15, 0.31, 0.665, 0.745),
yend = c(0, 0, 0.2, 0.2)) |>
ggplot(aes(x = x, xend = xend,
y = y, yend = yend)) +
geom_segment(arrow = my_arrow, color = pp[4]) +
annotate(geom = "text",
x = c(0.065, 0.31, 0.36, 0.64, 0.79), y = 0.5,
label = c("'~'", "'~'", "italic(j)", "'~'", "'~'" ),
size = c(10, 10, 7, 10, 10),
color = pp[4], family = "Times", parse = TRUE) +
xlim(0, 1) +
theme_void()
# Student-t density
p11 <- tibble(x = seq(from = -3, to = 3, by = 0.1)) |>
ggplot(aes(x = x, y = (dt(x, 3) / max(dt(x, 3))))) +
geom_area(fill = pp[9]) +
annotate(geom = "text",
x = 0, y = 0.2,
label = "student t",
color = pp[4], size = 7) +
annotate(geom = "text",
x = c(-1.4, 0), y = 0.6,
label = c("nu", "mu[italic(i)]"),
color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0)) +
theme_void() +
theme(axis.line.x = element_line(color = pp[4], linewidth = 0.5))
# Log sigma
p12 <- tibble(x = 0.65,
y = 0.6,
label = "log~sigma[italic(j)*'('*italic(i)*')']") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(hjust = 0, color = pp[4], family = "Times", parse = TRUE, size = 7) +
scale_x_continuous(expand = c(0, 0), limits = c(0, 2)) +
ylim(0, 1) +
theme_void()
# Two annotated arrows
p13 <- tibble(x = 0.15,
y = c(1, 0.47),
xend = c(0.15, 0.72),
yend = c(0.75, 0.1)) |>
ggplot(aes(x = x, xend = xend,
y = y, yend = yend)) +
geom_segment(arrow = my_arrow, color = pp[4]) +
annotate(geom = "text",
x = c(0.1, 0.15, 0.28), y = c(0.92, 0.64, 0.22),
label = c("'~'", "nu*minute+1", "'='"),
color = pp[4], family = "Times", parse = TRUE, size = c(10, 7, 10)) +
xlim(0, 1) +
theme_void()
# One annotated arrow
p14 <- tibble(x = 0.38,
y = 0.65,
label = "'='") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[4], family = "Times", parse = TRUE, size = 10) +
geom_segment(x = 0.5, xend = 0.5,
y = 1, yend = 0.15,
arrow = my_arrow, color = pp[4]) +
xlim(0, 1) +
theme_void()
# Another annotated arrow
p15 <- tibble(x = c(0.58, 0.71),
y = 0.6,
label = c("'~'", "italic(j)")) |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[4], family = "Times", parse = TRUE, size = c(10, 7)) +
geom_segment(x = 0.85, xend = 0.42,
y = 1, yend = 0.18,
arrow = my_arrow, color = pp[4]) +
xlim(0, 1) +
theme_void()
# The final annotated arrow
p16 <- tibble(x = c(0.375, 0.625),
y = c(1/3, 1/3),
label = c("'~'", "italic(i)")) |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[4], family = "Times", parse = TRUE, size = c(10, 7)) +
geom_segment(x = 0.5, xend = 0.5,
y = 1, yend = 0,
arrow = my_arrow, color = pp[4]) +
xlim(0, 1) +
theme_void()
# Some text
p17 <- tibble(x = 0.5,
y = 0.5,
label = "italic(y[i])") |>
ggplot(aes(x = x, y = y, label = label)) +
geom_text(color = pp[4], family = "Times", parse = TRUE, size = 7) +
xlim(0, 1) +
theme_void()
# Define the layout
layout <- c(
area(t = 1, b = 1, l = 8, r = 9),
area(t = 2, b = 3, l = 8, r = 9),
area(t = 3, b = 4, l = 3, r = 5),
area(t = 3, b = 4, l = 7, r = 9),
area(t = 3, b = 4, l = 11, r = 13),
area(t = 3, b = 4, l = 15, r = 17),
area(t = 6, b = 7, l = 1, r = 3),
area(t = 6, b = 7, l = 5, r = 9),
area(t = 6, b = 7, l = 11, r = 13),
area(t = 5, b = 6, l = 3, r = 17),
area(t = 10, b = 11, l = 6, r = 8),
area(t = 10, b = 11, l = 7, r = 9),
area(t = 8, b = 10, l = 1, r = 8),
area(t = 8, b = 10, l = 6, r = 8),
area(t = 8, b = 10, l = 6, r = 13),
area(t = 12, b = 12, l = 6, r = 8),
area(t = 13, b = 13, l = 6, r = 8))
# Combine, adjust, and display
(p1 + p2 + p3 + p4 + p5 + p6 + p7 + p8 + p9 + p10 + p11 + p12 + p13 + p14 + p15 + p16 + p17) +
plot_layout(design = layout) &
ylim(0, 1) &
theme(plot.margin = margin(0, 5.5, 0, 5.5))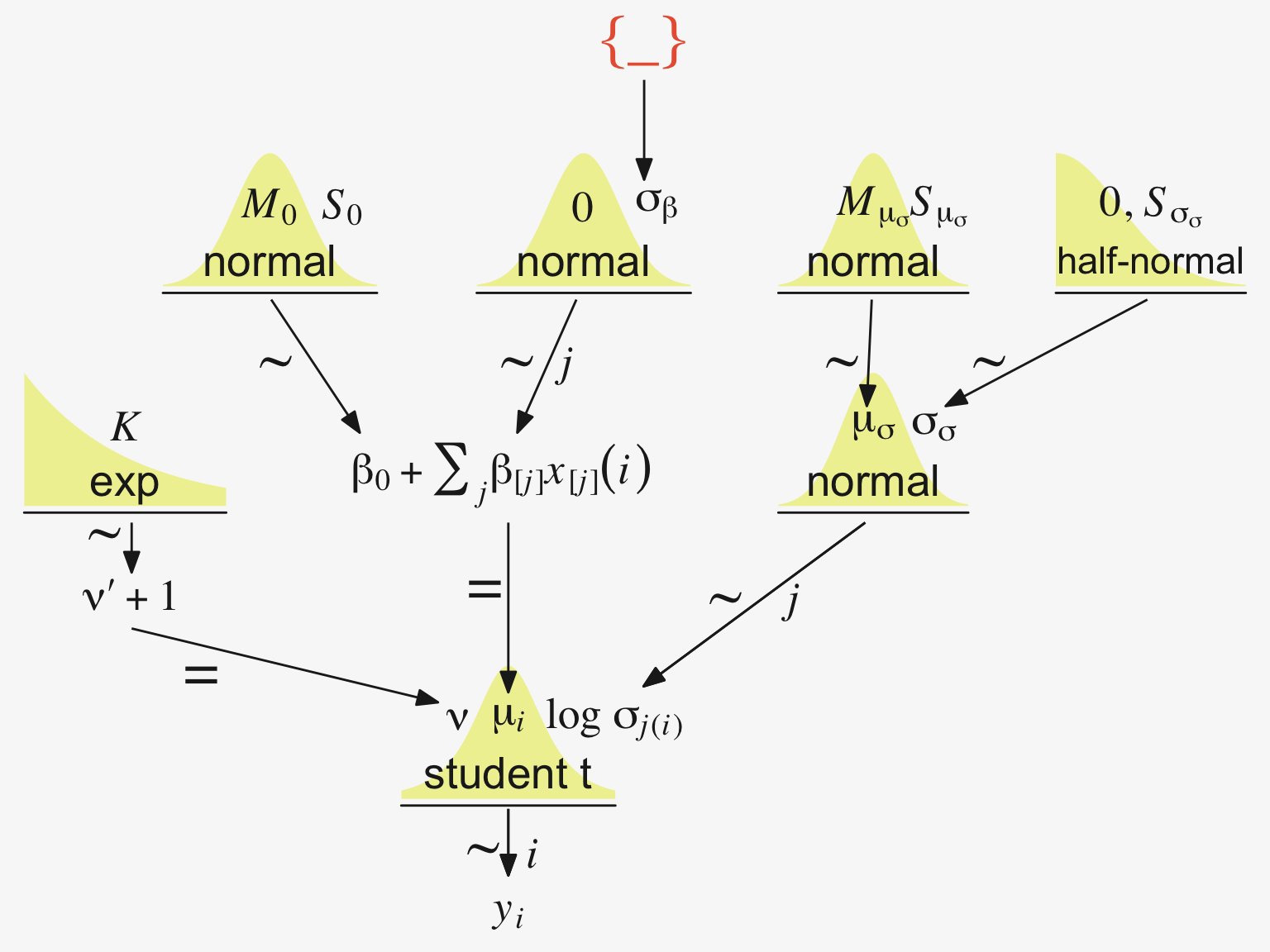
Since we’re modeling \(\log \left (\sigma_{j(i)} \right)\), we might use Gaussian prior centered on sd(my_data$y) |> log() and a reasonable spread like 1. We can simulate a little to get a sense of what those distributions look like.
n_draws <- 1e3
set.seed(19)
tibble(prior = rnorm(n_draws, mean = log(1), sd = 1)) |>
mutate(prior_exp = exp(prior)) |>
pivot_longer(cols = everything()) |>
ggplot(aes(x = value)) +
stat_dots(slab_color = pp[5], slab_fill = pp[5]) +
scale_y_continuous(NULL, breaks = NULL, expand = expansion(mult = c(0, 0.05)),
limits = c(0, NA)) +
facet_wrap(~ name, scales = "free")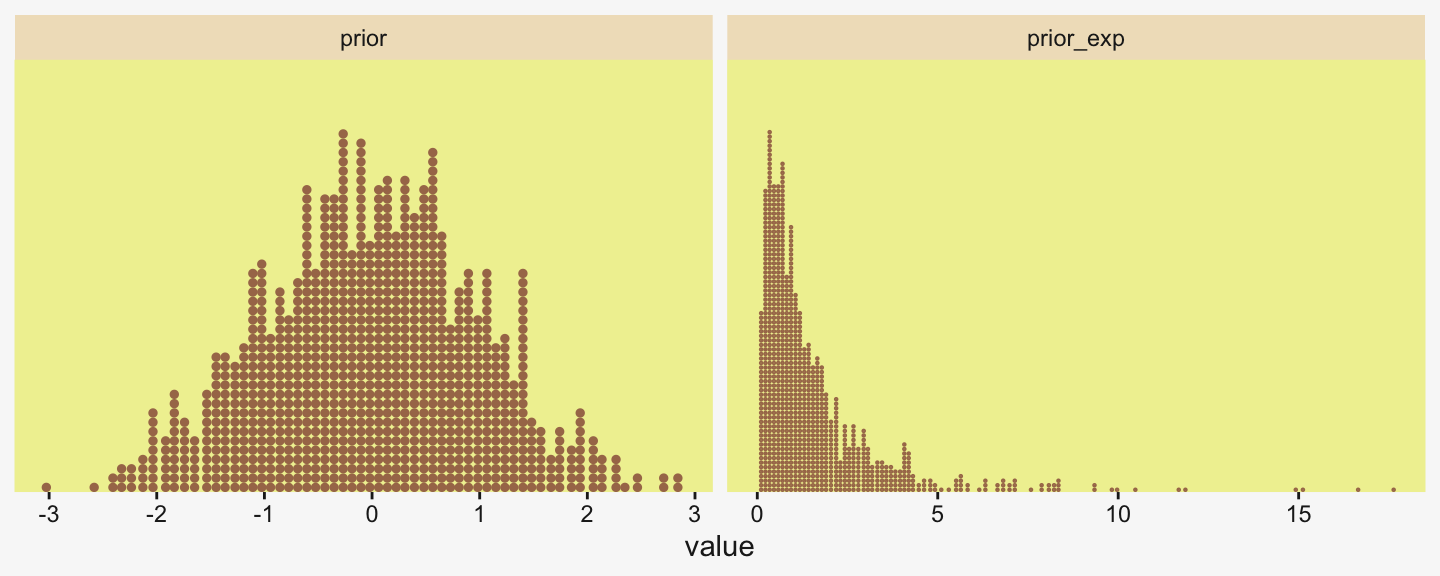
Here’s what is looks like with sd = 2.
set.seed(19)
tibble(prior = rnorm(n_draws, mean = log(1), sd = 2)) |>
mutate(prior_exp = exp(prior)) |>
ggplot(aes(x = prior_exp)) +
stat_dots(slab_color = pp[5], slab_fill = pp[5]) +
scale_x_continuous(expand = expansion(mult = c(0, 0.05))) +
scale_y_continuous(NULL, breaks = NULL, expand = expansion(mult = c(0, 0.05)),
limits = c(0, NA)) +
coord_cartesian(xlim = c(0, 17))
Though we’re still peaking around 1, there’s more mass in the tail, making it easier for the likelihood to pull away from the prior mode.
But all this is the prior on the fixed effect, the grand mean of \(\log (\sigma)\). Keep in mind we’re also estimating group-level deflections using a hierarchical model. The good old folded \(t\) on the unit scale is already pretty permissive for an estimate that is itself on the log scale. To make it more conservative, set \(\nu\) to infinity and go with a folded Gaussian. Or keep your regularization loose and go with a low-\(\nu\) folded \(t\) or even a folded Cauchy. And, of course, one could even go with a gamma.
Consider we have data my_data for which our primary variable of interest is y. Starting from preparing our stanvars values, here’s what the model code might look like.
# Get ready for `stanvars`
mean_y <- mean(my_data$y)
sd_y <- sd(my_data$y)
omega <- sd_y / 2
sigma <- 2 * sd_y
s_r <- gamma_a_b_from_omega_sigma(mode = omega, sd = sigma)
# Define `stanvars`
stanvars <- stanvar(mean_y, name = "mean_y") +
stanvar(sd_y, name = "sd_y") +
stanvar(s_r$shape, name = "alpha") +
stanvar(s_r$rate, name = "beta") +
stanvar(1/29, name = "one_over_twentynine")
# Fit the model
fit <- brm(
data = my_data,
family = student,
bf(Longevity ~ 1 + (1 | CompanionNumber),
sigma ~ 1 + (1 | CompanionNumber)),
prior = c(# Grand means
prior(normal(mean_y, sd_y * 5), class = Intercept),
prior(normal(log(sd_y), 1), class = Intercept, dpar = sigma),
# The priors controlling the spread for our hierarchical deflections
prior(gamma(alpha, beta), class = sd),
prior(normal(0, 1), class = sd, dpar = sigma),
# Don't forget our student-t nu
prior(exponential(one_over_twentynine), class = nu)),
stanvars = stanvars)19.5.1 Example: Contrast of means with different variances
Let’s load and take a look at Kruschke’s simulated group data.
my_data <- read_csv("data.R/NonhomogVarData.csv")
head(my_data)# A tibble: 6 × 2
Group Y
<chr> <dbl>
1 A 97.8
2 A 99.9
3 A 92.4
4 A 96.9
5 A 101.
6 A 80.7Here are the means and \(\textit{SD}\)s for each Group.
my_data |>
group_by(Group) |>
summarise(mean = mean(Y),
sd = sd(Y))# A tibble: 4 × 3
Group mean sd
<chr> <dbl> <dbl>
1 A 97 8.00
2 B 99 1.000
3 C 102 1.00
4 D 104. 8 First we’ll fit the model with homogeneous variances. To keep things simple, here we’ll fit a conventional model following the form of our original fit1. Here are our stanvars.
(mean_y <- mean(my_data$Y))[1] 100.5(sd_y <- sd(my_data$Y))[1] 6.228965omega <- sd_y / 2
sigma <- 2 * sd_y
(s_r <- gamma_a_b_from_omega_sigma(mode = omega, sd = sigma))$shape
[1] 1.283196
$rate
[1] 0.09092861# Define `stanvars`
stanvars <- stanvar(mean_y, name = "mean_y") +
stanvar(sd_y, name = "sd_y") +
stanvar(s_r$shape, name = "alpha") +
stanvar(s_r$rate, name = "beta")Now fit the ANOVA-like homogeneous-variances model.
fit19.6 <- brm(
data = my_data,
family = gaussian,
Y ~ 1 + (1 | Group),
prior = c(prior(normal(mean_y, sd_y * 10), class = Intercept),
prior(gamma(alpha, beta), class = sd),
prior(cauchy(0, sd_y), class = sigma)),
iter = 4000, warmup = 1000, chains = 4, cores = 4,
seed = 19,
control = list(adapt_delta = 0.995),
stanvars = stanvars,
file = "fits/fit19.06")Here’s the model summary.
print(fit19.6) Family: gaussian
Links: mu = identity
Formula: Y ~ 1 + (1 | Group)
Data: my_data (Number of observations: 96)
Draws: 4 chains, each with iter = 4000; warmup = 1000; thin = 1;
total post-warmup draws = 12000
Multilevel Hyperparameters:
~Group (Number of levels: 4)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(Intercept) 4.93 3.36 1.51 14.21 1.00 2024 2803
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 100.52 3.06 94.32 106.85 1.00 2045 1994
Further Distributional Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 5.76 0.44 4.98 6.70 1.00 6152 5832
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Let’s get ready to make our version of the top of Figure 19.7. First we wrangle.
# How many model-implied Gaussians would you like?
n_draws <- 20
densities <- my_data |>
distinct(Group) |>
add_epred_draws(fit19.6, ndraws = n_draws, seed = 19, dpar = c("mu", "sigma")) |>
mutate(ll = qnorm(0.025, mean = mu, sd = sigma),
ul = qnorm(0.975, mean = mu, sd = sigma)) |>
mutate(Y = map2(.x = ll, .y = ul,
.f = \(ll, ul) seq(from = ll, to = ul, length.out = 100))) |>
unnest(Y) |>
mutate(density = dnorm(Y, mean = mu, sd = sigma)) |>
group_by(.draw) |>
mutate(density = density / max(density))
glimpse(densities)Rows: 8,000
Columns: 12
Groups: .draw [20]
$ Group <chr> "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A",…
$ .row <int> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1…
$ .chain <int> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N…
$ .iteration <int> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N…
$ .draw <int> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1…
$ .epred <dbl> 98.34888, 98.34888, 98.34888, 98.34888, 98.34888, 98.34888, 98.34888, 98.34888, 98.34888, 98.34888, 98.34888,…
$ mu <dbl> 98.34888, 98.34888, 98.34888, 98.34888, 98.34888, 98.34888, 98.34888, 98.34888, 98.34888, 98.34888, 98.34888,…
$ sigma <dbl> 5.570203, 5.570203, 5.570203, 5.570203, 5.570203, 5.570203, 5.570203, 5.570203, 5.570203, 5.570203, 5.570203,…
$ ll <dbl> 87.43148, 87.43148, 87.43148, 87.43148, 87.43148, 87.43148, 87.43148, 87.43148, 87.43148, 87.43148, 87.43148,…
$ ul <dbl> 109.2663, 109.2663, 109.2663, 109.2663, 109.2663, 109.2663, 109.2663, 109.2663, 109.2663, 109.2663, 109.2663,…
$ Y <dbl> 87.43148, 87.65203, 87.87259, 88.09314, 88.31369, 88.53425, 88.75480, 88.97535, 89.19591, 89.41646, 89.63701,…
$ density <dbl> 0.1465288, 0.1582290, 0.1705958, 0.1836410, 0.1973740, 0.2118018, 0.2269281, 0.2427538, 0.2592764, 0.2764897,…In our wrangling code, the main thing to notice is those last two lines. If you look closely to Kruschke’s Gaussians, you’ll notice they all have the same maximum height. Up to this point, ours haven’t. This has to do with technicalities on how densities are scaled. In brief, the wider densities have been shorter. So those last two lines scaled all the densities within the same group to the same metric. Otherwise the code was business as usual.
Anyway, here’s our version of the top panel of Figure 19.7.
densities |>
ggplot(aes(x = Y, y = Group)) +
geom_ridgeline(aes(height = density, group = interaction(Group, .draw)),
color = adjustcolor(pp[7], alpha.f = 2/3), fill = NA,
linewidth = 1/3, scale = 3/4) +
geom_jitter(data = my_data,
alpha = 3/4, color = pp[10], height = 0.04) +
scale_x_continuous(breaks = seq(from = 80, to = 120, by = 10)) +
labs(y = NULL,
title = "Data with Posterior Predictive Distrib.") +
coord_cartesian(xlim = c(75, 125),
ylim = c(1.25, 4.5)) +
theme(axis.text.y = element_text(hjust = 0),
axis.ticks.y = element_blank())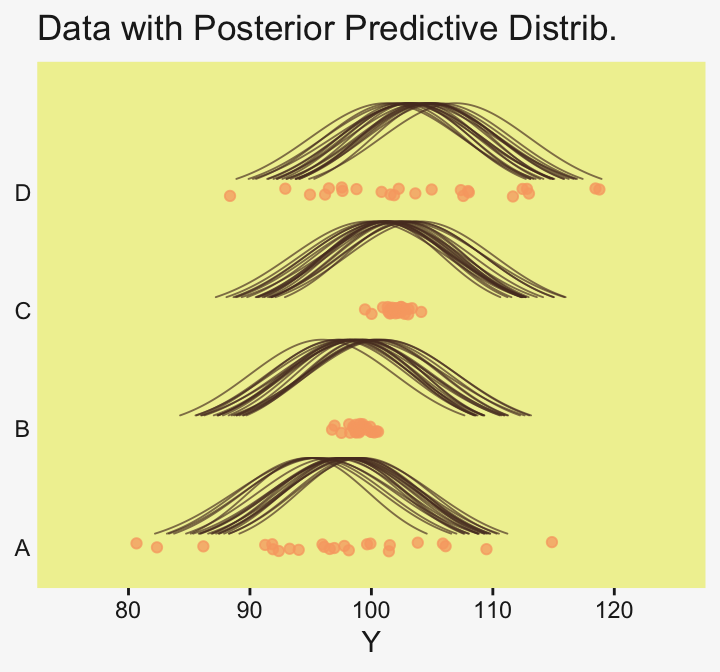
Here are the difference distributions in the middle of Figure 19.7.
draws <- as_draws_df(fit19.6)
differences <- draws |>
transmute(`D vs A` = `r_Group[D,Intercept]` - `r_Group[A,Intercept]`,
`C vs B` = `r_Group[C,Intercept]` - `r_Group[B,Intercept]`)
differences |>
pivot_longer(cols = everything()) |>
mutate(name = factor(name, levels = c("D vs A", "C vs B"))) |>
ggplot(aes(x = value)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_continuous(NULL, breaks = NULL) +
labs(x = "Difference") +
facet_wrap(~ name, scales = "free_x")
Now here are the effect sizes at the bottom of the figure.
differences |>
mutate_all(.funs = ~ . / draws$sigma) |>
pivot_longer(cols = everything()) |>
mutate(name = factor(name, levels = c("D vs A", "C vs B"))) |>
ggplot(aes(x = value)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_continuous(NULL, breaks = NULL) +
labs(x = "Effect Size (SMD)") +
facet_wrap(~ name, scales = "free_x")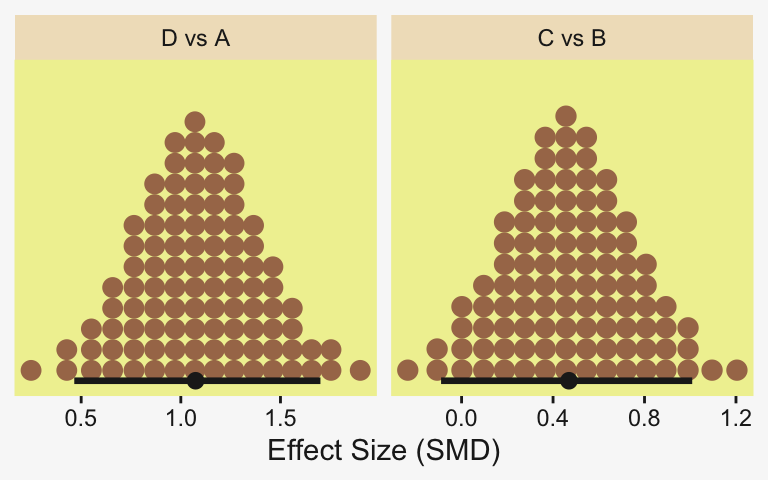
Oh and remember, if you’d like to get all the possible contrasts in bulk, tidybayes has got your back.
fit19.6 |>
spread_draws(r_Group[Group,]) |>
compare_levels(r_Group, by = Group) |>
# These next two lines allow us to reorder the contrasts along the y
ungroup() |>
mutate(Group = reorder(Group, r_Group)) |>
ggplot(aes(x = r_Group, y = Group)) +
geom_vline(xintercept = 0, color = pp[12]) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
labs(x = "Contrast",
y = NULL) +
coord_cartesian(ylim = c(1.5, 6.5)) +
theme(axis.text.y = element_text(hjust = 0),
axis.ticks.y = element_blank())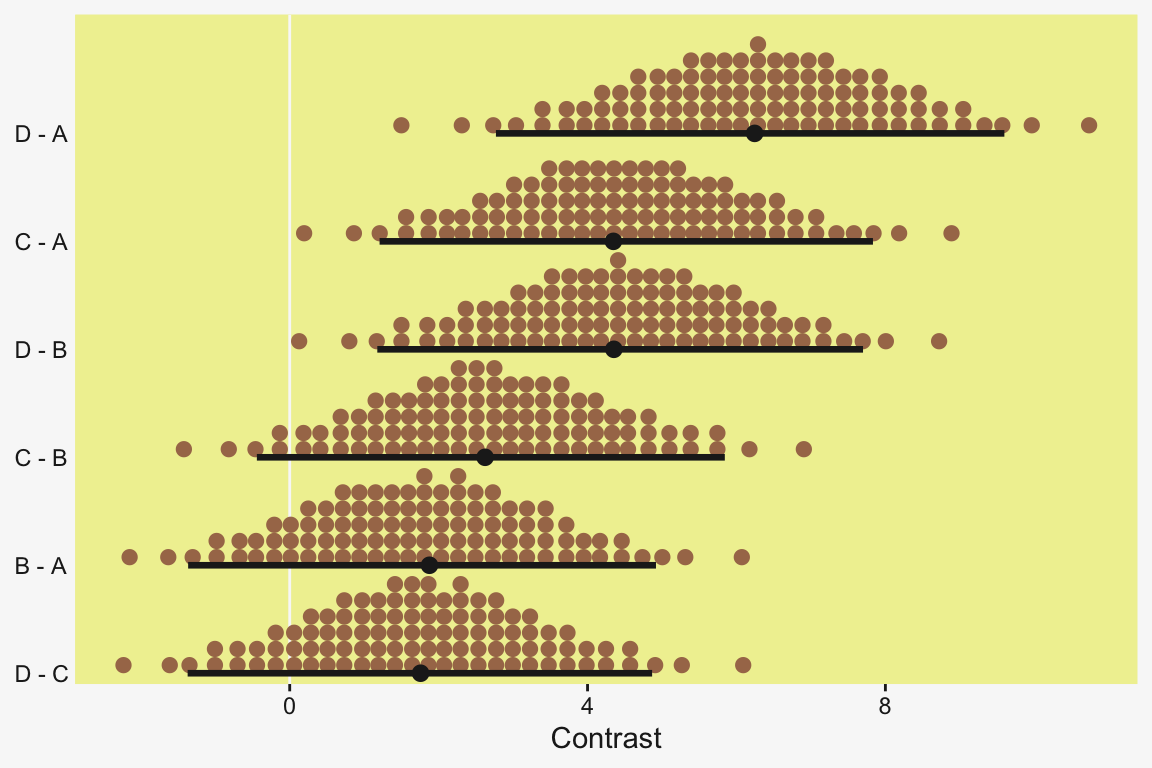
But to get back on track, here are the stanvars for the robust hierarchical variances model.
stanvars <- stanvar(mean_y, name = "mean_y") +
stanvar(sd_y, name = "sd_y") +
stanvar(s_r$shape, name = "alpha") +
stanvar(s_r$rate, name = "beta") +
stanvar(1/29, name = "one_over_twentynine")Now fit that robust better-than-ANOVA model.
fit19.7 <- brm(
data = my_data,
family = student,
bf(Y ~ 1 + (1 | Group),
sigma ~ 1 + (1 | Group)),
prior = c(# Grand means
prior(normal(mean_y, sd_y * 10), class = Intercept),
prior(normal(log(sd_y), 1), class = Intercept, dpar = sigma),
# The priors controlling the spread for our hierarchical deflections
prior(gamma(alpha, beta), class = sd),
prior(normal(0, 1), class = sd, dpar = sigma),
# Don't forget our student-t nu
prior(exponential(one_over_twentynine), class = nu)),
iter = 4000, warmup = 1000, chains = 4, cores = 4,
seed = 19,
control = list(adapt_delta = 0.99, max_treedepth = 12),
stanvars = stanvars,
file = "fits/fit19.07")The chains look good.
plot(fit19.7, widths = 2:3)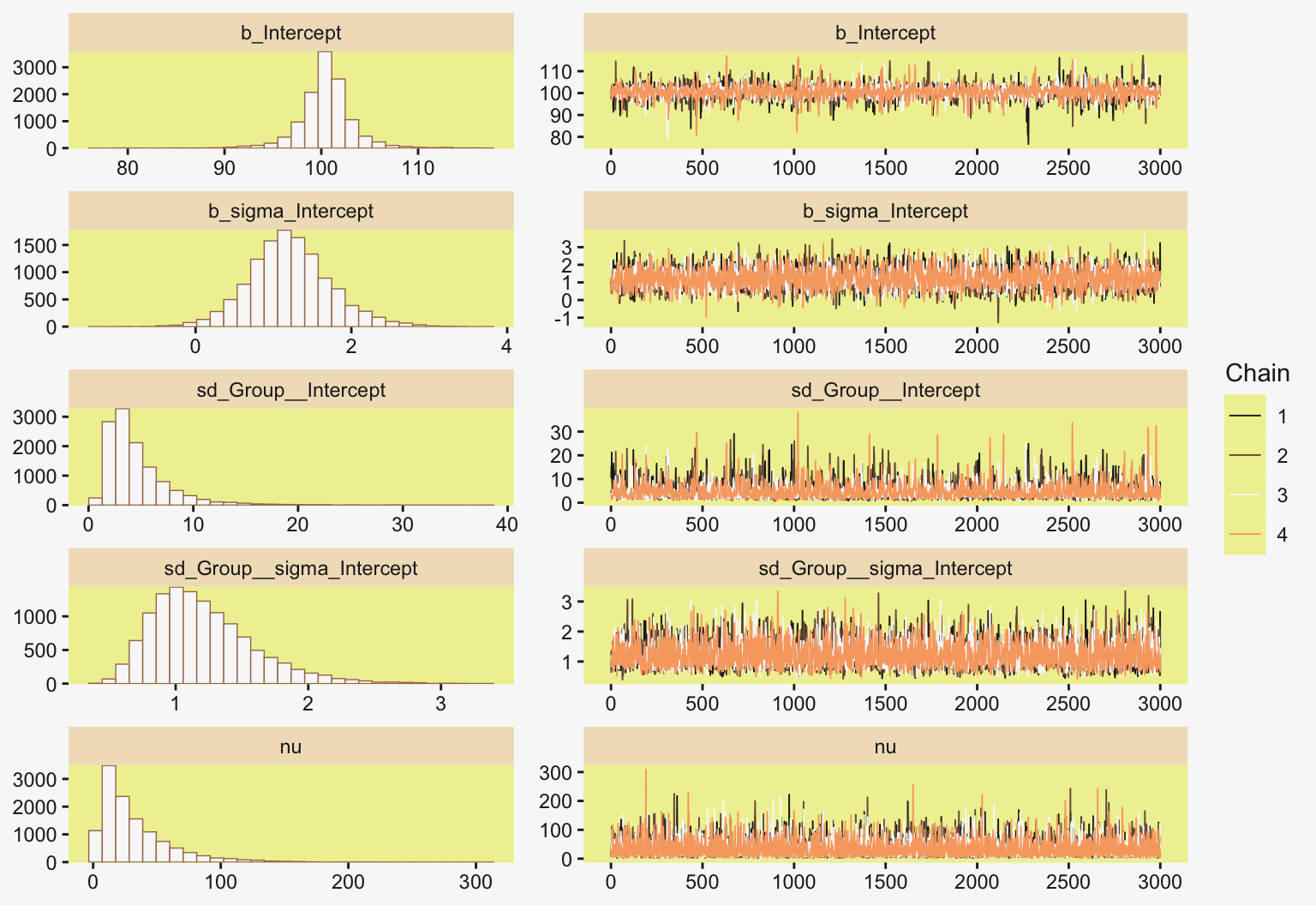
Here’s the parameter summary.
print(fit19.7) Family: student
Links: mu = identity; sigma = log
Formula: Y ~ 1 + (1 | Group)
sigma ~ 1 + (1 | Group)
Data: my_data (Number of observations: 96)
Draws: 4 chains, each with iter = 4000; warmup = 1000; thin = 1;
total post-warmup draws = 12000
Multilevel Hyperparameters:
~Group (Number of levels: 4)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(Intercept) 4.61 3.20 1.35 13.21 1.00 3244 5062
sd(sigma_Intercept) 1.21 0.39 0.64 2.15 1.00 4487 5023
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 100.49 2.82 94.60 106.22 1.00 4091 3762
sigma_Intercept 1.21 0.52 0.22 2.34 1.00 4236 5758
Further Distributional Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
nu 32.17 28.26 4.38 109.51 1.00 10070 7467
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Let’s get ready to make our version of the top of Figure 19.7. First we wrangle.
densities <- my_data |>
distinct(Group) |>
add_epred_draws(fit19.7, ndraws = n_draws, seed = 19, dpar = c("mu", "sigma", "nu")) |>
mutate(ll = qt(0.025, df = nu),
ul = qt(0.975, df = nu)) |>
mutate(Y = map2(.x = ll, .y = ul,
.f = \(ll, ul) seq(from = ll, to = ul, length.out = 100))) |>
unnest(Y) |>
mutate(density = dt(Y, df = nu)) |>
# Notice the conversion
mutate(Y = mu + Y * sigma) |>
group_by(.draw) |>
mutate(density = density / max(density))
glimpse(densities)Rows: 8,000
Columns: 13
Groups: .draw [20]
$ Group <chr> "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A",…
$ .row <int> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1…
$ .chain <int> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N…
$ .iteration <int> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, N…
$ .draw <int> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1…
$ .epred <dbl> 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 9…
$ mu <dbl> 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 95.8735, 9…
$ sigma <dbl> 5.250768, 5.250768, 5.250768, 5.250768, 5.250768, 5.250768, 5.250768, 5.250768, 5.250768, 5.250768, 5.250768,…
$ nu <dbl> 6.2418, 6.2418, 6.2418, 6.2418, 6.2418, 6.2418, 6.2418, 6.2418, 6.2418, 6.2418, 6.2418, 6.2418, 6.2418, 6.241…
$ ll <dbl> -2.424117, -2.424117, -2.424117, -2.424117, -2.424117, -2.424117, -2.424117, -2.424117, -2.424117, -2.424117,…
$ ul <dbl> 2.424117, 2.424117, 2.424117, 2.424117, 2.424117, 2.424117, 2.424117, 2.424117, 2.424117, 2.424117, 2.424117,…
$ Y <dbl> 83.14503, 83.40217, 83.65931, 83.91645, 84.17359, 84.43073, 84.68787, 84.94501, 85.20215, 85.45930, 85.71644,…
$ density <dbl> 0.09054714, 0.09720163, 0.10433744, 0.11198520, 0.12017662, 0.12894443, 0.13832222, 0.14834422, 0.15904513, 0…If you look closely at our code, above, you’ll note switching from the Gaussian to the Student \(t\) required changes in our flow. Most obviously, we switched from qnorm() and dnorm() to qt() and dt(), respectively. The base R Student \(t\) functions don’t take arguments for \(\mu\) and \(\sigma\). Rather, they’re presumed to be 0 and 1, respectively. That means that for our first three mutate() functions, the computations were all based on the standard Student \(t\), with only the \(\nu\) parameter varying according to the posterior. The way we corrected for that was with the fourth mutate().
Now we’re ready to make and save our version of the top panel of Figure 19.7.
p1 <- densities |>
ggplot(aes(x = Y, y = Group)) +
geom_ridgeline(aes(height = density, group = interaction(Group, .draw)),
color = adjustcolor(pp[7], alpha.f = 2/3), fill = NA,
linewidth = 1/3, scale = 3/4) +
geom_jitter(data = my_data,
alpha = 3/4, color = pp[10], height = 0.04) +
scale_x_continuous(breaks = seq(from = 80, to = 120, by = 10)) +
labs(y = NULL,
title = "Data with Posterior Predictive Distrib.") +
coord_cartesian(xlim = c(75, 125),
ylim = c(1.25, 4.5)) +
theme(axis.text.y = element_text(hjust = 0),
axis.ticks.y = element_blank())Here we make the difference distributions in the middle of Figure 19.8.
draws <- as_draws_df(fit19.7)
p2 <- draws |>
transmute(`D vs A` = `r_Group[D,Intercept]` - `r_Group[A,Intercept]`,
`C vs B` = `r_Group[C,Intercept]` - `r_Group[B,Intercept]`) |>
pivot_longer(cols = everything()) |>
mutate(name = factor(name, levels = c("D vs A", "C vs B"))) |>
ggplot(aes(x = value)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_continuous(NULL, breaks = NULL) +
xlab("Difference") +
facet_wrap(~ name, scales = "free_x")And here we make the plots of the corresponding effect sizes at the bottom of the Figure 19.8.
# First compute and save the sigma_j's, which will come in handy later
draws <- draws |>
mutate(sigma_A = exp(b_sigma_Intercept + `r_Group__sigma[A,Intercept]`),
sigma_B = exp(b_sigma_Intercept + `r_Group__sigma[B,Intercept]`),
sigma_C = exp(b_sigma_Intercept + `r_Group__sigma[C,Intercept]`),
sigma_D = exp(b_sigma_Intercept + `r_Group__sigma[D,Intercept]`))
p3 <- draws |>
# Note we're using pooled standard deviations to standardize our effect sizes, here
transmute(`D vs A` = (`r_Group[D,Intercept]` - `r_Group[A,Intercept]`) / sqrt((sigma_A^2 + sigma_D^2) / 2),
`C vs B` = (`r_Group[C,Intercept]` - `r_Group[B,Intercept]`) / sqrt((sigma_B^2 + sigma_C^2) / 2)) |>
pivot_longer(cols = everything()) |>
mutate(name = factor(name, levels = c("D vs A", "C vs B"))) |>
ggplot(aes(x = value)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_continuous(NULL, breaks = NULL) +
xlab(expression(Effect~Size~("Cohen's "*italic(d)[italic(t)[sigma]]))) +
facet_wrap(~ name, scales = "free_x")Combine them all and plot!
p1 / p2 / p3 + plot_layout(heights = c(2, 1, 1))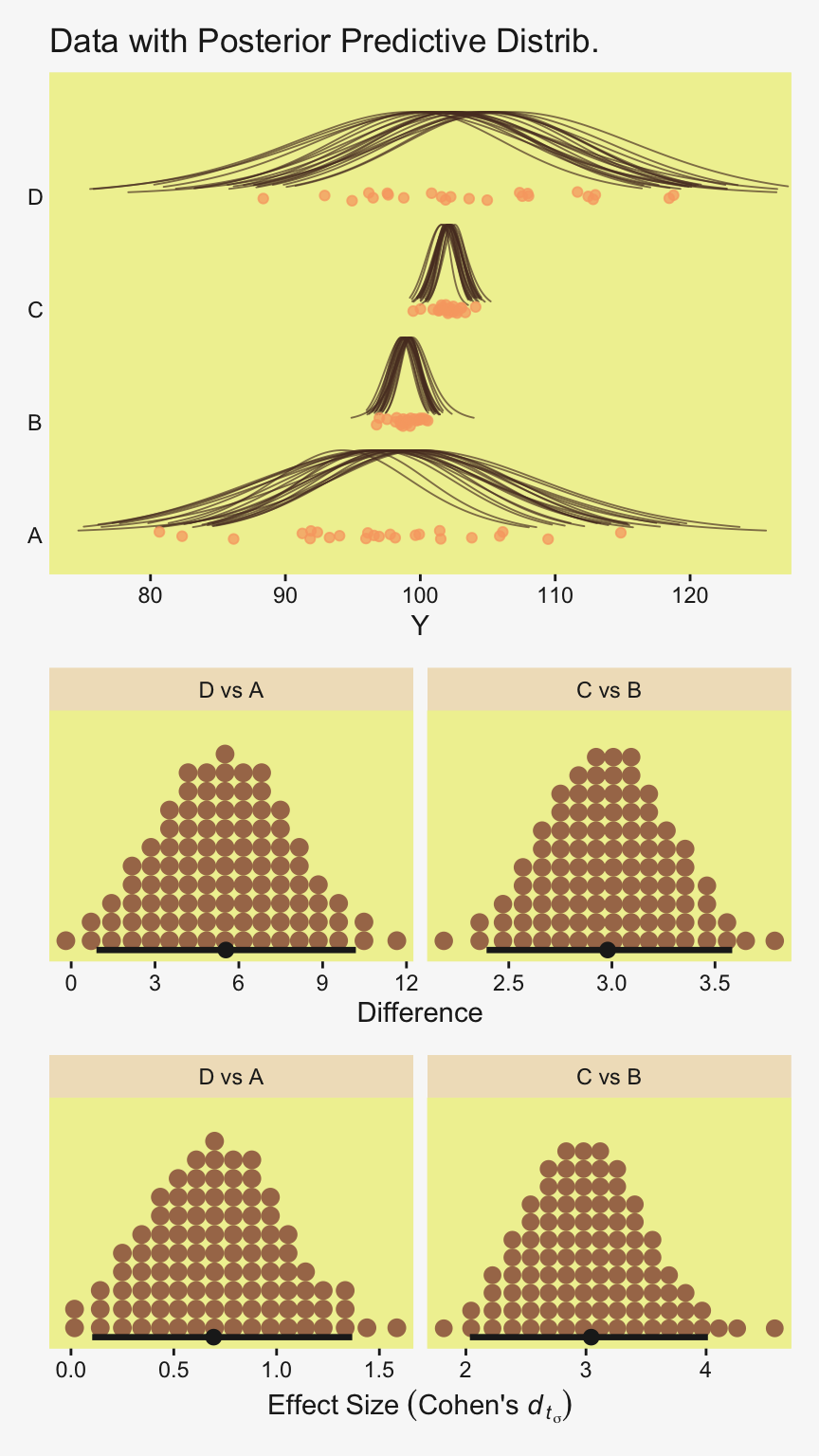
Notice that because (a) the \(\sigma\) parameters were heterogeneous and (b) they were estimated on the log scale, we had to do quite a bit more data processing before they effect size estimates were ready.
Also note that we have clarified, here, the effect size was a Cohen’s \(d_{t_{\sigma}}\). This notation is not common, but recall it’s what we discussed in Section 16.5.4 to distinguish between Cohen’s \(d\) SMDs from Student-\(t\) models standardized with a pooled \(\sigma\), versus those standardized with a pooled \(\textit{SD}\). Kruschke seems to like standardizing with a pooled scale (as displayed in the plot), whereas I generally prefer standardizing with a pooled \(\textit{SD}\).
Regarding the distinction between a Student-\(t\) scale and an \(\textit{SD}\), we might take a closer look at the \(\nu\) posterior.
draws |>
ggplot(aes(x = nu)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_x_continuous(expression(nu)) +
scale_y_continuous(NULL, breaks = NULL)
That \(\nu\) has a long uncertain right tail. But it also has a left tail coming in pretty close to the lower end, and we might take a closer look at the lowest few values.
draws |>
select(nu) |>
slice_min(nu, n = 4)# A tibble: 4 × 1
nu
<dbl>
1 1.84
2 2.03
3 2.07
4 2.08The lowest of the 12,000 posterior draws was just below the 2 threshold. As such, we cannot compute a pooled \(\textit{SD}\) for this model, and therefore cannot compute more conventional Cohen’s \(d_{t_{\textit{SD}}}\) effect sizes. If we were interested in them, we could refit the model using the lb = 2 setting for the prior of class = nu.1
1 Well okay, we could also compute a pooled \(\textit{SD}\) without refitting the model by using the rejection sampling method we briefly discussed in Section 16.5.2. For that method, we just drop all posterior draws for which nu <= 2.
“Finally, because each group has its own estimated scale (i.e., \(\sigma_j\)), we can investigate differences in scales across groups” (p. 578). That’s not a bad idea. Even though Kruschke didn’t show this in the text, we may as well give it a go.
# Recall we computed the sigma_j's a couple blocks up.
# Now we put them to use
draws |>
transmute(`D vs A` = sigma_D - sigma_A,
`C vs B` = sigma_C - sigma_B,
`D vs C` = sigma_D - sigma_C,
`B vs A` = sigma_B - sigma_A) |>
pivot_longer(cols = everything()) |>
mutate(name = factor(name, levels = c("D vs A", "C vs B", "D vs C", "B vs A"))) |>
ggplot(aes(x = value)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_y_continuous(NULL, breaks = NULL) +
expand_limits(x = 0) +
labs(x = expression(Differences~'in'~sigma[italic(j)]),
subtitle = expression("Just keep in mind "*sigma!=italic(SD)*" for the Student-"*italic(t))) +
facet_wrap(~ name, ncol = 4, scales = "free")
For more on models including a hierarchical structure on both the mean and scale structures, check out Donald Williams and colleagues’ work on what they call Mixed Effect Location and Scale Models [MELSM; e.g., Donald R. Williams et al. (2021); Donald R. Williams et al. (2019)]. They’re quite handy and I’ve begun using them in my applied work (e.g., here). You can also find a brief introduction to MELSM’s within the context of the multilevel growth model in Section 14.6 of my (2023) translation of McElreath’s (2020) second edition.
19.5.2 Bonus: Get those finite \(\textit{SD}_j\) posteriors
Let’s practice fitting another robust better-than-ANOVA model, but this time with the \(\nu > 2\) constraint to guarantee finite \(\textit{SD}_j\) posteriors.
fit19.7b <- brm(
data = my_data,
family = student,
bf(Y ~ 1 + (1 | Group),
sigma ~ 1 + (1 | Group)),
prior = c(
prior(normal(mean_y, sd_y * 10), class = Intercept),
prior(normal(log(sd_y), 1), class = Intercept, dpar = sigma),
prior(gamma(alpha, beta), class = sd),
prior(normal(0, 1), class = sd, dpar = sigma),
# This is the only update
prior(exponential(one_over_twentynine), class = nu, lb = 2)),
iter = 4000, warmup = 1000, chains = 4, cores = 4,
seed = 19,
control = list(adapt_delta = 0.98),
stanvars = stanvars,
file = "fits/fit19.07b")Check the model summary.
print(fit19.7b) Family: student
Links: mu = identity; sigma = log
Formula: Y ~ 1 + (1 | Group)
sigma ~ 1 + (1 | Group)
Data: my_data (Number of observations: 96)
Draws: 4 chains, each with iter = 4000; warmup = 1000; thin = 1;
total post-warmup draws = 12000
Multilevel Hyperparameters:
~Group (Number of levels: 4)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(Intercept) 4.49 3.08 1.32 12.79 1.00 3253 5432
sd(sigma_Intercept) 1.22 0.39 0.65 2.17 1.00 4471 5792
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 100.49 2.63 95.00 105.93 1.00 4578 4683
sigma_Intercept 1.22 0.53 0.22 2.35 1.00 4620 5744
Further Distributional Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
nu 32.01 28.03 4.49 106.34 1.00 8540 6326
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Here’s a plot comparing the two versions of the Cohen’s \(d_t\) effect sizes, by standardizer.
draws <- as_draws_df(fit19.7b) |>
# Compute group scales
mutate(sigma_A = exp(b_sigma_Intercept + `r_Group__sigma[A,Intercept]`),
sigma_B = exp(b_sigma_Intercept + `r_Group__sigma[B,Intercept]`),
sigma_C = exp(b_sigma_Intercept + `r_Group__sigma[C,Intercept]`),
sigma_D = exp(b_sigma_Intercept + `r_Group__sigma[D,Intercept]`)) |>
# Compute group SDs
mutate(sd_A = sigma_A * sqrt(nu / (nu - 2)),
sd_B = sigma_B * sqrt(nu / (nu - 2)),
sd_C = sigma_C * sqrt(nu / (nu - 2)),
sd_D = sigma_D * sqrt(nu / (nu - 2)))
draws |>
# Note we're using pooled standard deviations to standardize our effect sizes, here
transmute(.draw = .draw,
da_sd = (`r_Group[D,Intercept]` - `r_Group[A,Intercept]`) / sqrt((sd_A^2 + sd_D^2) / 2),
cb_sd = (`r_Group[C,Intercept]` - `r_Group[B,Intercept]`) / sqrt((sd_B^2 + sd_C^2) / 2),
da_sigma = (`r_Group[D,Intercept]` - `r_Group[A,Intercept]`) / sqrt((sigma_A^2 + sigma_D^2) / 2),
cb_sigma = (`r_Group[C,Intercept]` - `r_Group[B,Intercept]`) / sqrt((sigma_B^2 + sigma_C^2) / 2)) |>
pivot_longer(cols = -.draw) |>
separate_wider_delim(cols = name, delim = "_", names = c("contrast", "standarzider")) |>
mutate(contrast = factor(contrast, levels = c("da", "cb"), labels = c("D vs A", "C vs B")),
standarzider = factor(standarzider, levels = c("sd", "sigma"), labels = c("italic(SD)[italic(p)]", "sigma[italic(p)]"))) |>
ggplot(aes(x = value, y = standarzider)) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_x_continuous(expand = expansion(mult = 0.2)) +
scale_y_discrete("Standardizer", labels = ggplot2:::parse_safe, expand = expansion(mult = 0.1)) +
xlab(expression(Effect~Size~("Cohen's "*italic(d)[italic(t)["<Standardizer>"]]))) +
facet_wrap(~ contrast, scales = "free_x")
In this case because of the relatively high \(\nu\) posterior, the \(d_{t_\textit{SD}}\) effect sizes are only a bit smaller than Kruschke’s \(d_{t_\sigma}\) effect sizes. But they do differ, even so. Compute your effect sizes with care, friends.
19.5.3 Bonus: One can standardize pairwise, and overall
Kruschke didn’t highlight this in the text, but when you have 3 or more nominal groups, you have additional options on how you might standardize. To my eye, the convention (within the Gaussian context) is to generalize the two-group formula to pool across all of the groups
\[ \sigma_p = \sqrt{\frac{\sigma_1^2 + \sigma_2^2 + \dots + \sigma_k^2}{k}}, \]
where \(k\) is the number of groups. In the case of a four-group ANOVA, that would be
\[ \sigma_p = \sqrt{\frac{\sigma_1^2 + \sigma_2^2 + \sigma_3^2 + \sigma_4^2}{4}}. \]
Within the Gaussian context, this is the typical standardizer used to compute and analyze Cohen’s \(d\) effect sizes in meta-analyses.2 However, one can also compute a pairwise standardizer for each unique pair of groups, which is what I believe Kruschke did in the text. For example, say one wanted to focus on the comparison of groups 1 and 2, and the comparison of groups 3 and 4. Those pairwise pooled standard deviations could be computed as \(\sqrt{(\sigma_1^2 + \sigma_2^2)/2}\) for the first contrast and \(\sqrt{(\sigma_3^2 + \sigma_4^2)/2}\) for the second. To help differentiate this from the overall pooled standard deviation, we might call these \(\sigma_\textit{pp}\), the pairwise pooled scales.
2 Note that whereas here we are using notation that implies these quantities have been computed with the posterior distribution from a Bayesian model, one can also apply the structure of the equation to sample statistics, where \(s_p = \sqrt{(s_1^2 + s_2^2 + \dots + s_k^2)/k}\).
Recall that within the Gaussian context, \(\sigma\) is both a scale and the \(\textit{SD}\). But for the \(t\) distribution, they differ. Thus once you generalize to robust \(t\) models, you can further differentiate between pairwise pooled scales and pairwise pooled \(\textit{SD}\)s, which we might denote \(\textit{SD}_\textit{pp}\). To get a sense of all these possible standardizers, we plot using the focal groupings from above.
levels_vec <- c("sd_p", "sigma_p", "sd_pp_da", "sigma_pp_da", "sd_pp_cb", "sigma_pp_cb")
labels_vec <- c("italic(SD[p])", "sigma[italic(p)]", "italic(SD[pp])", "sigma[italic(pp)]", "italic(SD[pp])", "sigma[italic(pp)]")
draws |>
# Overall
mutate(sigma_p = sqrt((sigma_A^2 + sigma_B^2 + sigma_C^2 + sigma_D^2) / 4),
sd_p = sqrt((sd_A^2 + sd_B^2 + sd_C^2 + sd_D^2) / 4)) |>
# Pairwise
mutate(sd_pp_da = sqrt((sd_A^2 + sd_D^2) / 2),
sd_pp_cb = sqrt((sd_B^2 + sd_C^2) / 2),
sigma_pp_da = sqrt((sigma_A^2 + sigma_D^2) / 2),
sigma_pp_cb = sqrt((sigma_B^2 + sigma_C^2) / 2)) |>
pivot_longer(cols = sigma_p:sigma_pp_cb) |>
mutate(standardizer = factor(name, levels = levels_vec, labels = labels_vec) |>
fct_rev(),
pairwise = case_when(
str_detect(string = name, pattern = "pp_da") ~ "D and A",
str_detect(string = name, pattern = "pp_cb") ~ "C and B",
.default = "Overall") |>
fct_rev()) |>
ggplot(aes(x = value, y = standardizer)) +
stat_pointinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4]) +
scale_x_continuous("Standardizer parameter space", limits = c(0, NA)) +
scale_y_discrete("Standardizer", labels = ggplot2:::parse_safe) +
facet_grid(pairwise ~ "", scales = "free_y", space = "free_y") +
theme(strip.text.x = element_blank())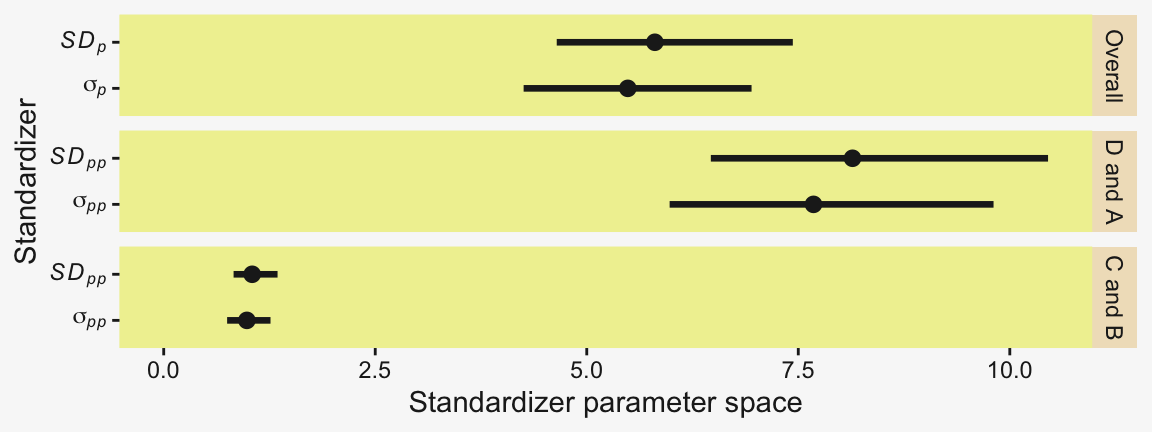
As expected, the \(\textit{SD}\) posteriors are systemically larger than their \(\sigma\) counterparts (compare them using the y axis). Thus any version of \(d\) standardized with a pooled \(\textit{SD}\) will be systemically smaller than its counterpart computed with a pooled \(\sigma\), which is a rehearsal from what we just saw in Section 19.5.2. But more importantly, look how much the pairwise standardizers for the D-A contrast (middle facet) differ from those for the C-B contrast (lower facet), and look how they both differ for the overall standardizers (upper facet). This means that the pairwise standardizers are on different metrics from one another, which effectively makes them uncomparable. To get a better sense of this, let’s look at another plot. For simplicity, this time we focus on the \(\sigma\)-based standardizers, though the overall point would be the same if we were to look at the \(\textit{SD}\)-based standardizers, instead.
levels_vec <- c("unit", "sigma_p", "sigma_pp")
labels_vec <- c("mu[1] - mu[2]", "(mu[1] - mu[2])/sigma[italic(p)]", "(mu[1] - mu[2])/sigma[italic(pp)]")
draws |>
# Mean contrasts
mutate(d_da = `r_Group[D,Intercept]` - `r_Group[A,Intercept]`,
d_cb = `r_Group[C,Intercept]` - `r_Group[B,Intercept]`,
# Standardizers
sigma_p = sqrt((sigma_A^2 + sigma_B^2 + sigma_C^2 + sigma_D^2) / 4),
sigma_pp_da = sqrt((sigma_A^2 + sigma_D^2) / 2),
sigma_pp_cb = sqrt((sigma_B^2 + sigma_C^2) / 2),
unit = 1) |>
pivot_longer(cols = c(d_da:d_cb, sigma_pp_da:sigma_pp_cb)) |>
mutate(name = str_remove(string = name, pattern = "pp_")) |>
separate_wider_delim(cols = name, delim = "_", names = c("quantity", "groups")) |>
pivot_wider(names_from = quantity, values_from = value) |>
rename(sigma_pp = sigma) |>
pivot_longer(cols = c(unit, sigma_p, sigma_pp), names_to = "standardizer") |>
mutate(groups = factor(groups, levels = c("cb", "da"), labels = c("C vs B", "D vs A")),
standardizer = factor(standardizer, levels = levels_vec, labels = labels_vec)) |>
ggplot(aes(x = d / value, y = groups)) +
geom_vline(xintercept = 0, color = pp[12]) +
stat_dotsinterval(point_interval = mode_hdi, .width = 0.95,
color = pp[4], quantiles = 100,
slab_fill = pp[5], slab_size = 0) +
scale_x_continuous("Difference", expand = expansion(mult = 0.2)) +
scale_y_discrete(NULL, expand = expansion(mult = 0.1)) +
facet_wrap(~ standardizer, scales = "free_x", labeller = label_parsed) 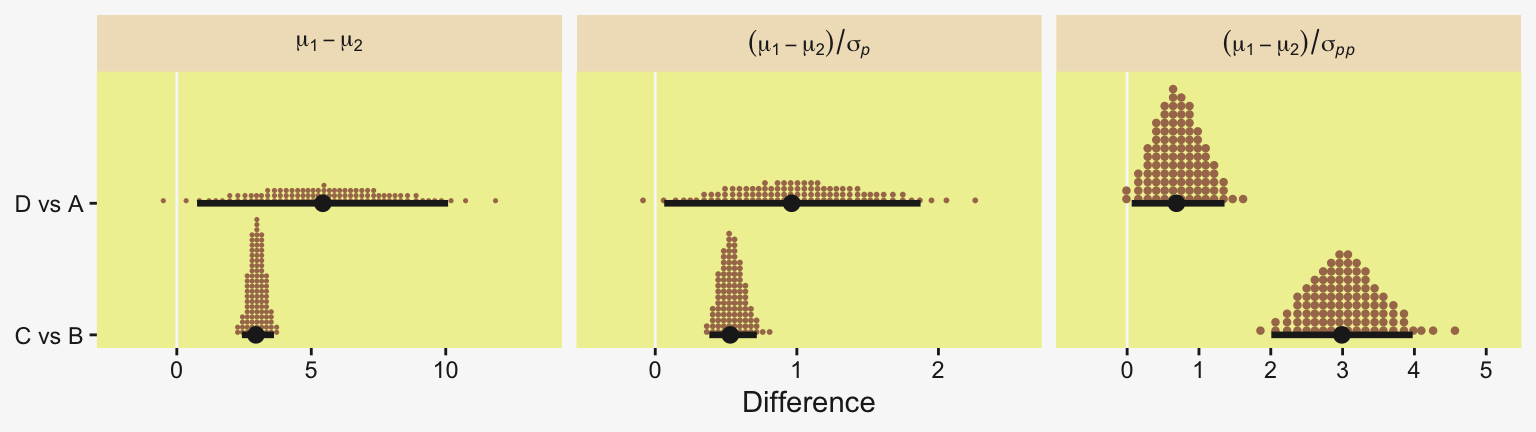
This time we use the y axis to highlight the two focal contrasts, D-vs-A and C-vs-B. In the facets, we showcase each contrast in three ways, by standardizer. The leftmost facet, \(\mu_i - \mu_2\), displays the unstandardized or raw mean differences, which is basically a re-expression of the differences in the middle row of Kruschke’s Figure 19.8. However, because they now share a common x axis, it’s easier to see the difference in their precision. The middle facet shows those contrasts expressed in a SMD standardized by the pooled scale, \(\sigma_p\). Were this a Gaussian model, the pooled scale would be the same as a pooled \(\textit{SD}\), which would be the conventional standardizer in most meta-analyses (or at least those I’ve seen). Notice that because the two focal contrasts share the same standardizer in the middle facet, their relative sizes are essentially the same as their unstandardized mean differences. All we have really done is rescale the x axis in the leftmost and middle facets.3
3 In addition to adding a little MCMC sampling error.
The rightmost facet shows the SMDs standardized by the pairwise pooled scales, \(\sigma_{pp}\). Notice that now the C-vs-B contrast appears to have a much larger value than the D-vs-A contrast, which is the opposite pattern from the other two facets. This discrepancy is due to the dramatic difference in their \(\sigma_{pp}\) values (look up again at the previous plot). But because the \(\sigma_{pp}\) values differ for the two focal contrasts, they are no longer directly comparable, even though we’ve given them a common x axis. If you compare these depictions to the bottom row of Kruschke’s Figure 19.8 in the text (p. 577), you’ll see the effect sizes he reported were computed with the \(\sigma_{pp}\), not the more conventional \(\sigma_p\).
Summing up: Because the pairwise pooled scale is different for each pairwise contrast, the contrasts are not directly comparable to one another; each is in its own metric. This is the opposite of the contrasts that used the pooled scale, which are all comparable to one another because they share a common standardizer. Thus when one of the research goals is to emphasize the differences in pairwise scales, Kruschke’s SMD with \(\sigma_\textit{pp}\) can be appropriate. But when researchers aim to compare the pairwise contrasts using a common metric, the more conventional \(\sigma_p\) standardizer may be preferred. This general pattern will hold when using pooled \(\textit{SD}\)s, rather than pooled scales.
19.5.3.1 Report your effect sizes effectively
There are many other ways to express effect sizes. Furthermore, \(d\)-type effect sizes aren’t appropriate for some model types or for some research questions. To expand your effect size repertoire, you might brush up on Cohen’s (1988) authoritative text or Cummings newer (2012) text. For nice conceptual overview on effect sizes, I recommend Kelley and Preacher’s (2012) paper, On effect size. Also see Pek and Flora’s (2018) handy paper, Reporting effect sizes in original psychological research: A discussion and tutorial.
Session info
sessionInfo()R version 4.5.1 (2025-06-13)
Platform: aarch64-apple-darwin20
Running under: macOS Ventura 13.4
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.5-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.5-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.1
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/Chicago
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] tidybayes_3.0.7 bayesplot_1.14.0 brms_2.23.0 Rcpp_1.1.0 patchwork_1.3.2 ggridges_0.5.7 palettetown_0.1.1
[8] lubridate_1.9.4 forcats_1.0.1 stringr_1.6.0 dplyr_1.1.4 purrr_1.2.0 readr_2.1.5 tidyr_1.3.1
[15] tibble_3.3.0 ggplot2_4.0.1 tidyverse_2.0.0
loaded via a namespace (and not attached):
[1] tidyselect_1.2.1 svUnit_1.0.8 viridisLite_0.4.2 farver_2.1.2 loo_2.8.0
[6] S7_0.2.1 fastmap_1.2.0 TH.data_1.1-4 tensorA_0.36.2.1 digest_0.6.37
[11] estimability_1.5.1 timechange_0.3.0 lifecycle_1.0.4 StanHeaders_2.32.10 survival_3.8-3
[16] magrittr_2.0.4 posterior_1.6.1 compiler_4.5.1 rlang_1.1.6 tools_4.5.1
[21] utf8_1.2.6 yaml_2.3.10 knitr_1.50 labeling_0.4.3 bridgesampling_1.1-2
[26] htmlwidgets_1.6.4 curl_7.0.0 pkgbuild_1.4.8 bit_4.6.0 plyr_1.8.9
[31] RColorBrewer_1.1-3 multcomp_1.4-29 abind_1.4-8 withr_3.0.2 numDeriv_2016.8-1.1
[36] grid_4.5.1 stats4_4.5.1 colorspace_2.1-2 inline_0.3.21 xtable_1.8-4
[41] emmeans_1.11.2-8 scales_1.4.0 MASS_7.3-65 cli_3.6.5 mvtnorm_1.3-3
[46] rmarkdown_2.30 crayon_1.5.3 generics_0.1.4 RcppParallel_5.1.11-1 rstudioapi_0.17.1
[51] reshape2_1.4.4 tzdb_0.5.0 rstan_2.32.7 splines_4.5.1 parallel_4.5.1
[56] matrixStats_1.5.0 vctrs_0.6.5 V8_8.0.1 Matrix_1.7-3 sandwich_3.1-1
[61] jsonlite_2.0.0 hms_1.1.4 arrayhelpers_1.1-0 bit64_4.6.0-1 ggdist_3.3.3
[66] glue_1.8.0 codetools_0.2-20 distributional_0.5.0 stringi_1.8.7 gtable_0.3.6
[71] QuickJSR_1.8.1 quadprog_1.5-8 pillar_1.11.1 htmltools_0.5.8.1 Brobdingnag_1.2-9
[76] R6_2.6.1 vroom_1.6.6 evaluate_1.0.5 lattice_0.22-7 backports_1.5.0
[81] rstantools_2.5.0 gridExtra_2.3 coda_0.19-4.1 nlme_3.1-168 checkmate_2.3.3
[86] xfun_0.53 zoo_1.8-14 pkgconfig_2.0.3
Comments